Article Text

Statistics from Altmetric.com
The gut associated immune system fences off potentially harmful intestinal antigens from the systemic circulation and induces systemic tolerance against luminal antigens. Intestinal immune responses against luminal antigens include IgA secretion and induction of regulatory cells. Unlike few other cytokines, lymphotoxin α/β regulates the development of intestinal lymphoid organs. The embryonic development of Peyer’s patches, postnatal lamina propria B cell development, and isolated lymphoid follicle development all depend on lymphotoxin β receptor interactions. Lymphotoxin α/β signalling also contributes to the development of mesenteric lymph nodes. In addition, intestinal inflammation is suppressed by inhibition of lymphotoxin β signalling, an observation which has initiated clinical studies using this treatment principal. Intestinal follicular lymphoid organs are sites of antigen presentation. Antigen presenting cells tune the delicate balance between intestinal immune tolerance and inflammation. Therefore, gut associated lymphatic organs and factors regulating their development are critical for the prevention of adverse immune reactions to intestinal antigens. This review provides an overview on the role of lymphotoxin and the gut associated lymphatic organs in the regulation of oral tolerance and intestinal inflammation.
INTRODUCTION
Intestinal mucosal surfaces are exposed to alimentary and bacterial antigens of the intestinal flora. The physiological immune response towards intestinal antigens is non-harmful to the entire organism and includes induction of systemic immune tolerance and IgA secretion. Inflammatory bowel disease (IBD) is associated with activation of the local intestinal and systemic immune responses. In various animal models of IBD, uncontrolled immune responses following intestinal injury result in mucosal insult. Colitis is associated with loss of tolerance against intestinal antigens, which also contributes to perpetuation of local and systemic inflammatory immune responses. Characterisation of intestinal inflammatory cytokine pathways has provided valuable tools to modulate the activity of IBD.
While inhibition of tumour necrosis factor receptor (TNFR) interactions is effective in controlling IBD, inhibition of interactions between the TNF family molecule lymphotoxin β(LTβ, bound in LTα1β2) with its receptor (LTβR) is currently being investigated as a potential IBD treatment. LTα1β2-LTβR interactions control the development and function of the intestinal immune system. During embryonic development, LTβR ligation is critical for the formation of gut associated lymphoid tissue (GALT). Postnatal LTα1β2-LTβR ligation controls the development of lamina propria (Lp) B cells. In adult mice, experimental colitis can be suppressed by inhibition of LTα1β2-LTβR signalling.
Presentation of antigens to immune effector cells is concentrated at sites of organised mucosal lymphoid follicles. The hallmark of organised GALT is the presence of lymphoid follicles. Therefore, GALT is the intestinal frontier of the systemic immune response. Figure 1 provides a schematic overview of the lymphoid organs of the intestinal immune system. As GALT organs are sites where antigen is presented to professional antigen presenting cells (APCs), they are critical for the decision between inflammation and tolerance. On the mucosal intestinal surface, only a thin layer of epithelial cells (IECs) separates the gut lumen from the mucosal immune system. Despite their barrier function, IECs provide a variety of transport functions to deliver information about the external milieu. Peyer’s patches (PP) are highly specialised “gut-type” lymphoid follicles in the small intestinal wall that contain naïve B cells, follicular dendritic cells (FDCs), and T cell rich areas. PPs are covered by the follicle associated epithelium (FAE) which contains specialised epithelial cells, called M cells. M cells sample luminal antigen directly into the PP, bypassing IECs. Isolated lymphoid follicles (ILF) and cryptopatches are additional intestinal lymphoid aggregates. ILFs are lymphoid aggregates in the antimesenteric wall of the small intestine which have been described in mice, humans,1 and other species. Colonic ILFs have also been reported in mice.2 Similar to PPs, ILFs consist of segregated B and T cell areas. They possess germinal centres and an overlying FAE containing M cells.2 Because of these structures, ILFs are suggested to be inductive sites for mucosal immune responses. Murine ILFs are up to 20 times more frequent than PPs and develop postnatally in a LTα1β2-LTβR dependent fashion.3

Schematic overview of the lymphoid elements of the gut associated lymphatic system. Peyer’s patches (PP) and mesenteric lymph nodes (MLN) are organised intestinal lymphoid follicles. (A–C) Pathways of intestinal antigen uptake: luminal antigen can be taken up by (A) intestinal epithelial cells, (B) interdigitating lamina propria dendritic cells, and by (C) M cells. The lymphatic drainage of PP and villus lamina propria goes to the MLNs (direction of lymph flow indicated by arrows).
-
Inhibition of interactions between the TNF family molecule lymphotoxin β with its receptor is currently being investigated as a potential IBD treatment.
Intestinal cryptopatches (CP) are small clusters of interleukin 7 receptor (IL-7R)+ lymphocytes in the Lp of the small and large intestine. There is substantial evidence indicating that gut CP develop progenitor T cells for extrathymic descendants which subsequently migrate to the intraepithelial lymphocyte (IEL) compartment.4–6 Colonic lymphoid follicles are present in the large intestine. IEL and Lp lymphocytes are distinct intestinal immune cell populations. Mesenteric lymph nodes (MLNs) provide a second line of defence as organised lymphoid tissue filtering the mesenteric lymph vessels.
Extraintestinal mucosal surfaces also develop specialised secondary lymphoid organs such as the nasal associated lymphoid tissue (NALT) and bronchus associated lymphoid tissue in the respiratory tract. Unlike PP, which develop during gestation, NALT organs develop after birth (reviewed by Mebius7).
Studies of the TNF cytokine family, especially LTα/β and its receptor, have provided a better understanding of factors regulating the development of the lymphoid GALT organs and Lp B lymphocytes. Furthermore, LTα1β2-LTβR interactions are involved in the control of intestinal inflammation. Investigators have thus been provided tools to differentially address the role of these organs in the regulation of intestinal tolerance and inflammation. This review provides an overview on recent studies covering the development of GALT organs, their role in experimental models of IBD and oral tolerance (OT), and the implications of LTα/β for the control of GALT organ formation and maintenance of an anti-inflammatory intestinal milieu.
STRUCTURE AND DEVELOPMENT OF GALT ORGANS
Development of secondary lymphoid organs in the intestine: lymphotoxin and IL-7 play a predominant role in GALT development
Overview and time course
Part of the organised GALT such as PPs seems to be genetically determined as they form at predictable sites during embryogenesis. Additionally, PPs accumulate in mice and human as they age. Colonic patches appear in disease (IBD).8 However, the triggers for this postnatal accumulation of intestinal secondary lymphoid tissues are not well understood. The development of PPs and MLNs is controlled by various cytokines and chemokines. Table 1 summarises gene deficient mice with defects in GALT development. Lymph nodes (LN) develop from lymph sacs by endothelial cell budding from veins, which are subsequently colonised by connective tissue. Lymph sacs give rise to the lymphatic vasculature by sprouting of lymphatic vessels (reviewed by Mebius7). MLNs are the first to develop in embryogenesis (gestational days 10.5–15.5), indicating a pivotal role of this organ for the entire organism. However, PPs develop late in gestation (gestational days 16–21). Intestinal CPs, intestinal Lp B cells, and NALT organs develop postnatally.7Figure 1 provides an overview on the different GALT organs.
Mice with defects in organised gut associated lymphoid tissue development induced by gene defects or by gestational or postgestational treatment (modified from Mebius7)
Central role of TNF/lymphotoxin for GALT development
TNF family cytokines, especially LT and the LTβ receptor (LTβR), play a critical role in the development of secondary lymphoid organs. LT-α forms soluble homotrimers (LTα3) and heterodimers (LTα1β2, LTα2β1) with the membrane molecule LTβ. LTα3 similar to TNF binds to the TNF receptors I (55 kDa) and II (75 kDa). LIGHT is another ligand of LTβR. Figure 2 depicts the different LT molecules and their receptors. LTα1β2 is expressed on haematopoietic cells, including T cells, B lymphocytes, and natural killer (NK) cells. The various biological effects of LTα1β2 are mediated by interaction of these circulating cells with LTβR, which is expressed on resident mesenchymal and stromal cells.9 LTα1β2-LTβR interactions are completely or partially disrupted in mice with targeted deletion of the respective cytokine and receptors genes (LTα gene deficient (−/−), LTβ−/−, LTβR−/− mice), or in mice lacking LTαβ expressing cell populations (µMT mice without mature B cells). Competitive inhibition of LTβR binding by soluble LTβR-IgG fusion protein (LTβRIgG) is another effective means of blocking LTβR signalling. Figure 3 depicts the biological effects of LTβR mediated signalling and its inhibition.
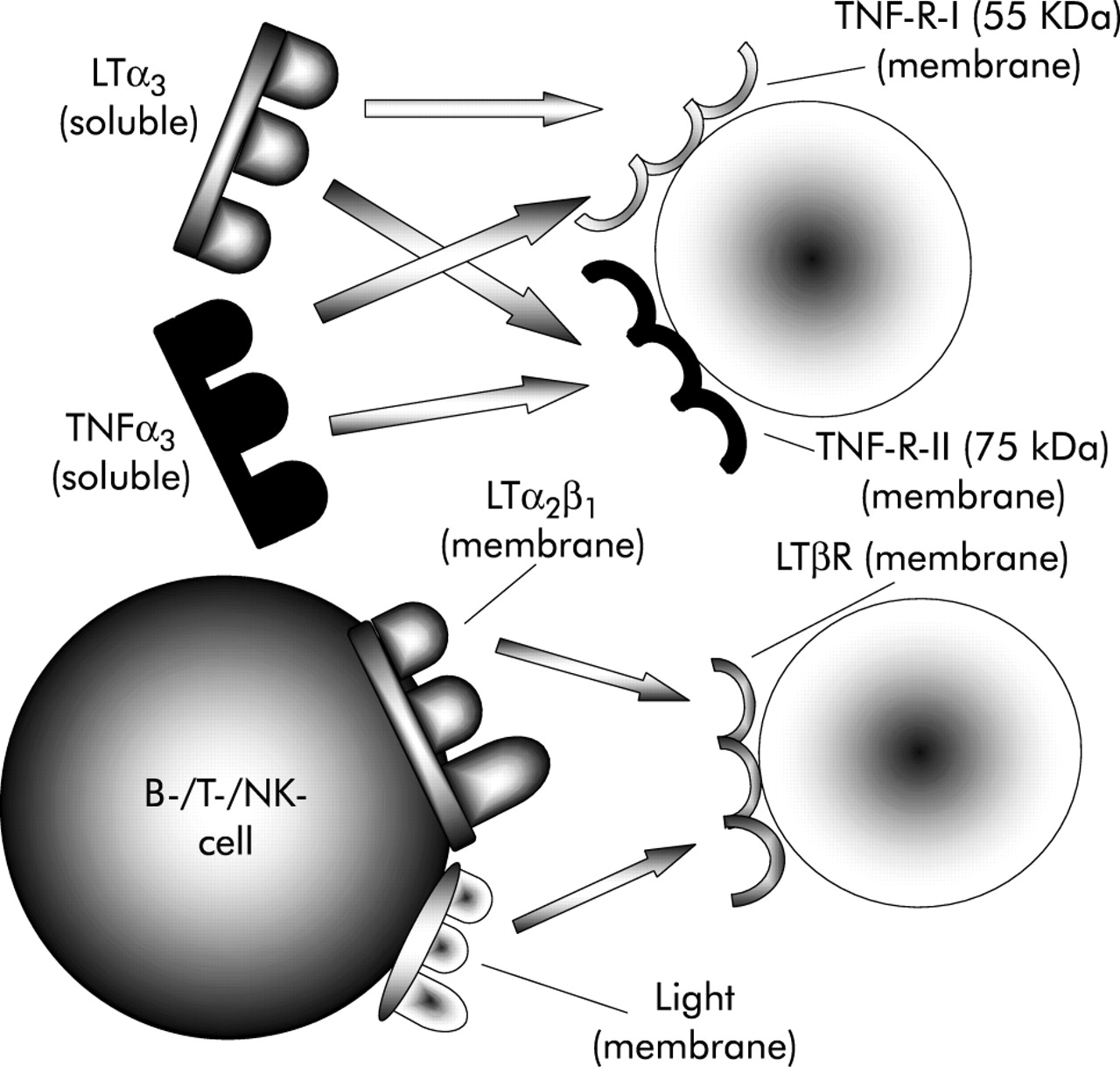
Schematic overview of LTα1β2-LTβR ligand receptor interactions. Soluble LTα3 interacts with the TNF receptors I (55 kDa) and II (75 kDa) similar to soluble TNFα3. LTα1β2 heterodimers and LIGHT are ligands for the membrane bound LTβR which is expressed on mesenchymal cells. LTα/β, -R, lymphotoxin α/β, -receptor; NK, natural killer; TNF, tumour necrosis factor; TNF-R, TNF receptor.

Biological effects of lymphotoxin (LT)α1β2-LTβR interaction. LTα1β2 heterodimers expressed on CD3−CD4+CD45+ cells (lymph node (LN) and Peyer’s patch (PP) development) or on B cells (lamina propria (Lp) B cell development), T cells, and natural killer cells interact with LTβR expressed on mesenchymal cells (LN and PP development) or Lp stroma cells (Lp B cell development) and induce signalling, as indicated (+ sign). Expression of LTα1β2 on CD3−CD4+CD45+ cells is upregulated by ligation of the interleukin 7 receptor (IL-7R) with interleukin 7 (IL-7). LTβR mediated signalling is induced (as indicated by a + sign) by ligation of LTβR with agonistic antibody (mAb) AF.H6 and inhibited by soluble LTβR- IgG fusion protein (IgG) (as indicated by a − (minus) sign). The tumour necrosis factor family cytokine LIGHT is a transmembrane molecule expressed on T cells which binds to LTβR. LIGHT signalling through LTβR is not required for PP formation but contributes to MLN formation in a LTα1LTβ2 dependent fashion. Black arrows indicate effects of LTβR mediated signalling on gut associated lymphoid tissue development and intestinal inflammation. Ligation of LTβR is critical for PP and LN development, formation of small intestine isolated lymphoid follicles (ILF), and recruitment of B cells into the Lp compartment. LTβR mediated signalling is involved in the regulation of intestinal inflammation as LTβRIgG treatment abrogated transfer colitis.
The role of IL-7 in GALT development
An early event in LN and PP formation is the occurrence of interleukin 7 receptor (IL-7R)+ CD3−CD4+CD45+ cells, which are progenitors of APCs and NK cells. Ligation of IL-7R and of TNF related activation induced cytokine (TRANCE) induces LTα1β2 expression on these cells.9–12 IL-7R+ cells colocalise with vascular cell adhesion molecule (VCAM)-1+ cell clusters. LTα1β2-LTβR interaction induces VCAM expression which can be inhibited by soluble IL-7R or blockade of LTα1β2-LTβR signalling pathways during embryogenesis.13 Ablation of IL-7 or of the IL-7 signalling pathway results in mice deficient in PP.13–16 Transgenic expression of IL-7 in the intestine of IL-7 gene deficient (−/−) mice restored PP formation.17 Gestational treatment of mice with anti-IL-7Rα antibody by a single injection on day 15.5 post-conception abrogated PP formation in progeny of treated mice.18
GALT defects secondary to inhibition of LTα1β2-LTβR and TNF-RI (55 kDa) signalling
Signalling through the LTβR by LTα1β2 is critical for PP development (fig 2). Disruption of LTα/β heterodimer formation in LTα−/− mice results in loss of PP and of all LN.19,20 Gestational inhibition of LTα1β2-LTβR interactions inhibits PP formation.21–23 Combined inhibition of LTβR and TNF-R-I (55) receptor-ligand interactions prevents the formation of PP and most LN.24 Conversely, stimulation of LTβR in LTα−/− mice by agonist anti-LTβR antibodies (fig 3) induces formation of MLN.24 LTβR−/− mice develop a phenotype similar to that observed in LTα−/− mice.25 Figure 4 provides a schematic overview on the effects of gestational blockade or stimulation of LTβR on the formation of GALT organs.

Effect of gestational treatment with soluble cytokine receptor fusion proteins (tumour necrosis factor receptor (TNF-R)-I (55 kDa)), lymphotoxin β receptor-IgG fusion protein (LTβRIgG), and agonist anti-LTβR antibody treatment on the formation of gut associated lymphoid tissue tissues. (A) Combined inhibition of TNF-R-I- and LTβR signalling by gestational intravenous treatment of TNF-R-I (55 kDa) and LTβRIgG, 10–4 days prior to birth, abrogates formation of Peyer’s patches (PP) and most lymph nodes (LNs) (PP null/LN null) in progeny of treated mothers. (B) Selective abrogation of PP formation (PP null/LN+) in wild-type mice is mediated by injection of mice with LTβRIgG 5–3 days prior to birth and within 24 hours after birth. (C) Formation of mesenteric lymph nodes (mLN) but not of PP (PP−/mLN+) is induced in progeny of PP and LN deficient LTα−/− mice by gestational treatment with agonist anti-LTβR antibody AF.H6 on days 10–4 prior to birth.
Interactions between LTα1β2 and LTβR are critical for recruitment of B cells into the Lp. There is dramatically reduced IgA secretion in LTα−/−,19,20 LTβ−/−,26 and LTβR−/−25 mice. Reconstitution of irradiated LTα−/− mice with wild-type bone marrow (BM) restored IgA secretion in BM chimera while PP/LN defects persisted.27,28 Thus PP and MLN are not required for IgA secretion. Post-gestational inhibition of LTα1β2-LTβR interactions inhibits migration of B cells into the Lp.28 Conversely, reconstitution of irradiated LTα−/− mice with wild-type or RAG2−/− BM restores the Lp B cell population28 and IgA secretion.27 LTβR expression on Lp stromal cells is crucial for restoration of B cell recruitment and IgA production. Expression of LTα1β2 on B cells contributes to PP formation as inhibition of B cell maturation in µMT mice impairs the development of PP.29
Signalling through the TNF-R-I also contributes to PP development as TNF-R-I (55)−/− mice develop small PP.30,31
ILFs are small intestinal lymphoid follicles consisting of T and B cell areas and an overlying FAE containing M cells.2,32 The role of these follicles in intestinal immune regulation are as yet unknown. In the human small intestine, there are also isolated lymphoid structures with an epithelium resembling the FAE, MHC class II+ dendritic cells (DC), memory T cells, and B cells but no immature lymphocytes.1 Human inflammatory bowel disease is associated with induction of secondary lymphoid follicles in the colon8 resembling ILFs. In ulcerative colitis (UC) there is lymphoid follicular hyperplasia of so-called basal lymphoid aggregates.33 These follicles are characterised by abnormal follicular architecture and unusual immunophenotypes with an increased proportion of apoptosis resistant cells and CD4−CD8−γδ T cell receptor α(TCR)+ cells. Lorenz et al demonstrated that the formation of ILFs depends on interactions between LTα1β2 and LTβR on non-BM derived cells and intact TNF-RI function.3
Other cytokines and chemokines involved in GALT development and maintenance
In addition to LTα1β2 and IL-7, LIGHT, a second ligand of the LTβR, also contributes to GALT organ formation.34 CXC chemokine ligand 13 (CXCL13, B lymphocyte attractant, BCL) and its ligand, CX chemokine receptor 5 (CXCR5), control the entry of B cells into LNs.35–39 T cell recruitment into lymphoid tissues is also controlled by CC chemokine receptor 7 (CCR7)-CC chemokine ligand 19 (CCL19) interactions. Another important chemokine is macrophage inflammatory protein 3α(CCL20) which is expressed by the FAE and can be induced under inflammatory conditions. CCL20 is the only known chemokine ligand for CCR6. CCL20 has chemotactic activity for B and T cell subpopulations and myeloid DCs that bear CCR6.40 The highly specific interaction of CCL20 and CCR6 and the predominant expression of CCL20 in the FAE suggest that this receptor-ligand pair may be involved in maintenance of organised MALT. Indeed, we recently observed morphological abnormalities in PP from CCR6−/− mice and a marked reduction in M cells in these mice (own unpublished observations). Most recently it was shown that regulated IEL development depends on CCR6 expression in lymphoid precursors in intestinal cryptopatches.41
Additional genes and cytokines/chemokines relevant for the organisation of secondary lymphoid tissues are summarised in table 1.
In summary, gestational development of organised GALT organs is predominantly controlled by TNF family cytokines and IL-7 under a tight time frame. Yet poorly defined factors contribute to the increase in PPs after birth. ILF development and Lp B cell recruitment while being regulated by the same factors develop after birth, suggesting a higher priority of MLNs and PPs than ILFs and Lp B cells in the evolutionary schedule. While LTα1β2-LTβR and IL-7 are major regulators for PP development and important contributors to MLN formation, TRANCE and LIGHT are dispensable for GALT organ formation. Homing of T and B cells to PPs and MLNs is controlled by CCR7-CCL19 and CXCL13-CXCR5 interactions.
PROFESSIONAL APCS IN ORGANISED GALT ORGANS
Both PP and MLN consist of T and B cell areas which are infiltrated by distinctive sets of DCs. In general, DCs are distinguished as myeloid CD8α−CD11b+ and lymphoid CD8 α+CD11b−. PP contain a unique set of CD8α−CD11b−DC. CD8α−CD11b−DC and CD8α−CD11b+ cells are present in the dome of PP.42–44 CD8α−CD11b+ PP DC predominantly secrete IL-10. Ligation of PP DC with receptor activator of nuclear factor κB (NFκB)(RANK) induces IL-10 secretion while splenic DC respond with IL-12 secretion.45 Similarly, PP DC are prone to induce T helper (TH2) responses in antigen specific T cells and receptor activator of NFκB ligand (RANK-L) stimulation enhances OT.44,45 MLN DCs most likely migrate from mucosal surfaces to the MLN. Secretion of IL-10 and transforming growth factor β(TGF-β) by MLN DC and stimulation of antigen specific CD4+ T cells for IL-10 and TGF-β have recently been demonstrated in mice following oral antigen.46,47
THE ROLE OF GALT ORGANS IN INTESTINAL INFLAMMATION: IBD IS FOCUSED AT SITES OF HIGHLY CONCENTRATED LYMPHOID TISSUE
Appendix in intestinal inflammation
There is a striking focus of intestinal inflammation in the ileocaecal region, a site with highly concentrated aggregation of secondary lymphoid tissue. In humans, ileocaecal colitis is common in IBD, such as Crohn’s disease (CD) or backwash ileitis in UC. However, infectious agents such as Yersinia enterocolitica and Mycobacteria also cause pseudoappendicitis and mesenteric lymphadenitis.48,49 The terminal ileum is also a preferential site of inflammation in several animal models of IBD.50,51 Circumstantial evidence indicates that the appendix plays a pivotal role in the development of CD and UC. There is a negative association between appendectomy and the onset and severity of these two diseases.52,53 Appendiceal inflammation is observed in half of colectomy specimens from patients with UC. The appendiceal epithelium shows intense upregulation of HLA class II molecules and activation of macrophages54 in specimens from UC, but not in acute appendicitis. Thus the appendix might serve as a priming site for UC.
-
Gestational development of organised GALT organs is predominantly controlled by TNF family cytokines and IL-7 under a tight time frame.
T cell receptor α chain gene deficient (TCRα−/−) mice develop spontaneous colitis which is similar to human UC.55 Cells in the appendix associated lymphoid follicle (ALF) proliferate and B cells secrete autoantibodies against tropomyosin.55 In humans, tropomyosin might also act as an autoantigen. In UC, there is also increased secretion of antibodies against tropomyosin.56 In TCRα−/− mice there was more B cell proliferation and a higher level of autoantibody production in ALF tissue than in PP tissue.55 Appendectomy at a young age inhibited the development of colitis in TCRα−/− mice and diminished the number of cells in MLN.55
There is severe colitis in both IL-2−/− and IL-2Rα chain−/− mice, which is similar to human UC.57,58 Half of all IL-2−/− mice die within the first nine weeks after birth after developing splenomegaly, lymphadenopathy, and autoimmune anaemia. One hundred per cent of surviving animals develop pancolitis with thickening of the entire colon during the next 6–15 weeks. Similar to colitis in TCRα−/− mice, there are autoantibodies which are directed against colonic antigens.
Lymphoid hyperplasia is a feature of chronic dextran sodium sulphate (DSS) induced colitis.59 The severity of acute DSS induced colitis was reduced in mice which had undergone appendectomy prior to disease induction.60 Thus two different animal disease models provide consistent data indicating that the appendix might play a pivotal role in the induction of experimental IBD.
Table 2 summarises studies on the role of GALT organs in experimental intestinal inflammation.
Studies using animal models with mice deficient of organised gut associated lymphoid tissue (GALT) organs and/or lymphotoxin α/β pathway
-
There is a striking focus of intestinal inflammation in the ileocaecal region, a site with highly concentrated aggregation of secondary lymphoid tissue.
Role of LTα1β2-LTβR interactions, lymphoid follicles, PP, and MLN in the development and outcome of murine colitis models: blockade of LTβR signalling can abrogate experimental colitis
Experimental colitis induced by transfer of TH1-type CD45RBhi cells or reconstitution of Tgϵ26 mice with wild-type BM is attenuated by LTβRIgG treatment during disease induction.61 The effect of LTβRIgG treatment on these two models of TH1 mediated colitis was similar to that of anti-TNF treatment. As a consequence of this study, anti-LTβ directed treatment of CD is currently being investigated.
A more recent study reported the course of TNBS colitis in mice made deficient of PP and colonic lymphoid follicles secondary to gestational inhibition of LTα1β2-LTβR interactions.62 Mice deficient in colonic patches developed focal ulcers with TH1-type responses whereas lesions in normal mice were of a diffuse mucosal type with both TH1- and TH2-type cytokine production.62 Thus colonic lymphoid follicles and PP provide help to control intestinal inflammation in trinitrobenzene sulphonic acid (TNBS) induced colitis.
Treatment of adult mice with LTβRIgG prior to disease induction and following the TNBS enema was associated with loss of PP and a reduction in DCs in the colon. Interferon γ−/− mice undergoing such treatment were protected from TNBS colitis. Peri-inductional treatment of wild-type mice with LTβRIgG induced hypertrophy of colonic patches following the TNBS enema. Thus TH2-type colitis is dependent on the presence of colonic patches or of LTα1LTβ2-LTβR interactions.
Thus both TH1-type and TH2-type cytokine mediated types of experimental colitis can be controlled by inhibition of the LTα1β2-LTβR pathway although the mechanism may vary in different disease models.
Data currently available indicate that inhibition of LTβR signalling suppresses intestinal inflammation by altering the local intestinal and splenic lymphoid microarchitecture. There are fewer DCs in colonic follicles and in the spleen following LTβRIgG treatment.62,63 Inhibition of LTβR signalling reduces the number and size of follicular DCs, germinal centres, and marginal zones in colonic follicles, all of which are important to mount full scale B cell responses.62 Figure 5 provides an overview of the known and potential mechanisms modulating intestinal inflammation by inhibition of LTβR signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Known and potential mechanisms of systemic inhibition of lymphotoxin receptor (LTβR) signal transduction resulting in control of colitis. (A) Loss of follicular dendritic cells (DC), B cell follicles, and DC content in colonic lymphoid follicles results in down modulation of local intestinal immune response. Lamina propria (Lp) B cells, T cells, and natural killer cells expressing LTα1β2 are inhibited in their contact with LTβR expressing Lp stromal cells and might thus be dysfunctional. (B) Loss of follicular DC, B cell follicles, and DC content in mesenteric lymph nodes (MLN) is possible following lymphotoxin β receptor-IgG fusion protein (LTβRIgG) treatment and might results in immunosuppression. (C) Blood borne intestinal antigens are carried to the spleen. LTβRIgG treatment diminishes the content of splenic DC and disturbs splenic architecture resulting in loss of ability to present antigen (bacterial antigens, T cell independent antigen type II).
We have recently studied acute DSS induced colitis in mice without PP and in mice lacking PP and MLN.64 We found more severe colitis in mice deficient in PP and LN (PP/LN null), in both LTα−/− mice and in PP/LN null mice secondary to gestational treatment with LTβRIgG and TNFRIgG.64 There was hyperplasia and induction of colonic lymphoid follicles in LTα−/− mice and PP/LN null mice, suggesting an LT independent pathway of formation of colonic lymphoid tissue induced during colonic inflammation. Colonic lymphoid patches observed in LTα−/− mice showed ill defined T and B cell areas while colonic follicles of wild-type PP/LN null mice showed distinct T and B cell zones in the colonic follicles, predominantly consisting of B cells. Clinically, DSS induced colitis was similar in wild-type and mice without PP but with MLN (PP null/LN+ mice), although there was also induction of colonic lymphoid tissue in PP null/LN+ mice. Thus PPs alone do not regulate DSS induced colitis whereas the presence of MLN is critical in this regard. The differences observed using mice with GALT defects are most likely due to the different animal models and strains used.
M cells are specialised epithelial cells of the FAE of PP. M cells have a high capacity for transcytosis of microorganisms and macromolecules and serve as an antigen sampling system (reviewed by Kucharzik and colleagues65). Experimental ileitis in the rat induces the formation of M cells, which subsequently undergo apoptosis.66
-
Data currently available indicate that inhibition of LTβR signalling suppresses intestinal inflammation by altering the local intestinal and splenic lymphoid microarchitecture.
THE ROLE OF LYMPHOTOXIN AND GALT ORGANS IN INTESTINAL IMMUNE TOLERANCE: MLN ARE CRITICAL AND PP MIGHT BE REDUNDANT FOR ORAL TOLERANCE
Immune tolerance is defined as induction of “any mechanism by which a potentially injurious immune response is prevented, suppressed, or shifted to a non-injurious class of immune response”.67 OT is a mechanism of tolerance induction, and OT has been demonstrated by the specific suppression of cellular and/or humoral immune responses to an antigen by prior administration of the antigen by the oral route. OT probably serves as a mechanism to prevent the development of adverse immune reactions against intestinal and nutritional antigens. The site/s of intestinal antigen presentation for OT induction is still not clearly defined. Table 3 summarises studies on the role of GALT organs in the induction of OT. IECs are the frontier of antigen contact and present antigen in vitro to CD8+ T cells.68,69 Such IEC activated CD8+ T cells have regulatory properties70 but their in vivo function for OT remain obscure. γδ TCR+ IEL play a role in induction of OT as TCR δ chain gene deficient mice and mice treated with anti-γδ-TCR antibodies have a defect in induction of OT.71–73µMT mice are deficient in mucosal B cells and can be orally tolerised.46 LTβ−/−, LTα+/−/LTβ+/−, and µMT mice deficient in PP can be orally tolerised.46,74 Thus the absence of LTβ itself does not abrogate induction of OT. Surgical removal of PP in rats failed to abrogate OT.75 Gestational treatment of mice with LTβRIgG renders progeny of treated mice deficient in PP. If such PP null mice were generated by intravenous treatment on gestational days 14 and 17, tolerance induction was abrogated.76 It is of note that such treatment similarly abrogated suppression of delayed type hypersensitivity, proliferative, total IgG responses, and TH1- and TH2-type responses against OVA, suggesting induction of a TH0-type immune response. OT to haptens was not abrogated in this study. However, in PP null mice generated by intravenous LTβRIgG treatment on days 16 and 18, OT was intact.74,77 The different observations indicate that the time course of gestational LTβRIgG treatment is critical to regulate different, yet poorly defined, pathways of intestinal immune responses, resulting in maintenance or loss of OT. Lp and/or MLN DCs which are prone to prime for regulatory T cells might be one potential target of early gestational LTβ inhibition. Postpartal treatment with LTβRIgG has been shown to inhibit colonic FDC and DC development.62 Further work will be necessary to address these unresolved questions.
Studies investigating the role of gut associated lymphoid tissue (GALT) organs in induction of intestinal tolerance and immunity
LTα−/− and TNF/LTα−/− mice without PP and MLN and Balb/c mice without PP and MLN (PP/LN null mice) secondary to gestational treatment of pregnant mice with LTβRIgG and TNFRIgG are resistant to induction of OT.74,77 However, peripheral immune tolerance could be induced by the intraperitoneal route in all PP/LN deficient mice used in these studies, suggesting that the defect in OT is situated at the GALT. Gestational signalling at the LTβR by treatment with agonistic anti-LTβR antibody AF.H6 induces formation of MLN in LTα−/− mice without inducing PP formation. OT was restored in progeny of AF.H6 treated LTα−/− mice, suggesting that the lack of MLN but not of PP and of the cytokine LTα is critical for OT induction.77
Intestinal DCs might play a pivotal role in induction of OT. We assume that intraluminal antigens that pass the intestinal epithelial barrier are taken up by DCs and then migrate through draining lymph vessels to MLN, where they present antigen to T cells. Alpan and colleagues46 reported priming of naive T cells for TH2 immune responses by MLN derived DC. Oral antigen was administered to in vivo pulsed MLN derived DC from mice with and without PP. In contrast, DC from popliteal LN failed to prime for TH2 T cell responses following subcutaneous footpad antigen administration. Rescigno and colleagues78 reported that DC penetrate gut epithelial monolayers to sample bacteria and might thus provide an M cell independent pathway for intestinal antigen uptake and presentation. It is possible that the lack of OT observed in mice without PP and MLN 74,77 is also related to a LTβR dependent block of recruitment of CD4+CD3− inducer cells, DC, or other cells to the MLN anlage or the Lp compartment, which are required for intestinal tolerance induction.
More recent studies by Blumberg (personal communication) indicate a third pathway of antigen transport through the intestinal epithelial cell barrier using the intestinal neonatal Fc receptor (FcRn) as a shuttle service. Antigen bound to IgG can be transported through IECs from the basolateral to the apical side and vice versa. Luminal antigen transported to the basolateral side of IECs primes MLN and hepatic APCs to induce antigen specific T cell responses.
-
OT probably serves as a mechanism to prevent the development of adverse immune reactions against intestinal and nutritional antigens.
SUMMARY: A MODEL FOR GALT ORGANS IN INTESTINAL INFLAMMATION AND TOLERANCE
Recent studies have shed new light on the mechanisms underlying the development of the organised GALT. TNF family cytokine members are among the master regulators of intestinal lymphoid development. Gestational LTα1LTβ2-LTβR interactions are critical for the development of PP whereas blockade of the LTα1LTβ2-LTβR pathway in adolescence is pivotal for Lp B cell development.
GALT organs are the intestinal branch of the organised lymphoid system. PP are sites of IgA production and thus contribute to local and systemic immunity against intestinal antigens. PPs with the FAE and M cells provide a pathway for uptake and processing of particulate antigens. As tolerance to soluble antigens could be induced in various gene deficient mice without PP and with MLN, PP are probably redundant for induction of OT. More likely soluble antigen is predominantly taken up by IECs, interdigitating Lp DCs and the FcRn to shuttle through the intestinal barrier. Antigen primed Lp DCs then migrate through the intestinal lymph vessels to MLNs. Antigen presentation in MLNs causes tolerance under physiological conditions. Tolerance is mediated by the induction of regulatory TH3-type79 cells and Treg cells,80 probably by secreting TGF-β and IL-10. Different mechanisms involved in tolerance induction include anergy and deletion of antigen specific cells. Intestinal inflammation is associated with loss of the immunological balance between pro- and anti-inflammatory cytokines. Interestingly, both TH1-type and TH2-type cytokines are potent mediators of intestinal inflammation, as indicated by the TNBS and oxazolone models of colitis. There is evidence for control of both inflammatory conditions by TGF-β. The change in the intestinal cytokine milieu in IBD contributes to induction of inflammatory T cell responses by Lp DC cells migrating to the MLN. We assume that MLNs serve as a site where DCs prime T cells for anti-inflammatory T cell responses as the course of acute DSS colitis was more severe in mice without MLNs than in those with MLNs. Human IBD and experimental colitis in mice are associated with immune activation in all GALT organs. Similar to MLNs, the appendix and the ALF seem to be critical for the development of both IBD and experimental colitis in animals as appendectomy attenuates both conditions.
-
Intestinal DCs might play a pivotal role in induction of OT.
-
GALT organs are the intestinal branch of the organised lymphoid system.
Inhibition of LTα1β2-LTβR signalling in TH1 and TH2 mediated experimental colitis suppresses intestinal inflammation by mechanisms yet poorly defined.
Future studies are necessary to address the mechanisms by which intestinal inflammation is down modulated following post-gestational inhibition of the LTα1β2-LTβR pathway.
Acknowledgments
This review is dedicated to Professor Wolfram Domschke on the occasion of his 60th birthday.