Article Text

Statistics from Altmetric.com
- tropical calcific pancreatitis
- fibrocalculous pancreatic diabetes
- cationic trypsinogen gene
- pancreatic secretory trypsin inhibitor gene
- ICP, idiopathic chronic pancreatitis
- HP, hereditary pancreatitis
- TCP, tropical calcific pancreatitis
- FCPD, fibrocalculous pancreatic diabetes
- CFTR, cystic fibrosis transmembrane regulator
- ERCP, endoscopic retrograde cholangio-pancreaticography
- GTT, glucose tolerance test
- PRSS1, protease, serine, 1 (trypsin 1)
- PSTI, pancreatic secretory trypsin inhibitor
- SPINK1, serine protease inhibitor, Kazal type I
Pancreatitis is a global health care problem with varied aetiologies. Alcoholism is responsible in the majority of patients while other causes, such as heredity, gallstones, hyperlipidaemia, hypercalcaemia, and idiopathic pancreatitis, are relatively rare.1,2 The causal factor in 20-30% of such cases is still not known and they fall into the category of idiopathic chronic pancreatitis (ICP).1,2 Although the exact pathogenesis is not clear, autodigestion secondary to aberrant intraductal activation of zymogens by trypsin is a primary common event. A genetic basis was reported in 1996 by familial linkage analysis3–5 and confirmed by detection of missense mutations, namely R122H and N29I, in the cationic trypsinogen gene (PRSS1) in hereditary pancreatitis (HP) patients.6,7 HP is a relatively rare autosomal dominant disorder where typical acute attacks in childhood and frequent progression to chronic pancreatitis are seen to occur in two or more subjects or generations. Subsequent studies from other parts of the world have also reported the two common mutations8,9 and other mutations in the PRSS1 gene in both HP and ICP patients.8,10,11 However, only about 60% cases of HP and less than 20% with a diagnosis of ICP have a mutated PRSS1 gene, suggesting the presence of other candidate genes.
Pancreatic secretory trypsin inhibitor (PSTI/SPINK1) is a potent protease inhibitor and thought to be a major protective mechanism preventing inappropriate activation of pancreatic digestive enzyme cascade by inhibiting up to 20% of potential trypsin activity.12 The human SPINK1 gene on chromosome 5 is approximately 7.5 kb long with four exons and codes for a product of 79 amino acids including a signal peptide of 23 amino acids.13 Since the inhibitor molecule provides the first line of defence against prematurely activated trypsinogen within the pancreas, it has recently attracted attention as a possible cause of chronic pancreatitis. A causal role for the SPINK1 gene was ruled out initially,14 but a significant correlation between mutated SPINK1 and chronic pancreatitis was first shown by Witt et al15 and subsequently supported by Pfutzer et al.16 A founder mutation N34S was detected in the majority of patients, while another variant P55S was identified in only a few. In contrast, two recent analyses have reported a very low prevalence of SPINK1 gene mutations. Five (three with N34S) out of 32 Japanese patients17 and only two (one with N34S) out of 20 German adult patients with idiopathic chronic pancreatitis18 had a mutated SPINK1 gene.
Tropical calcific pancreatitis (TCP) is an idiopathic, juvenile, non-alcoholic form of chronic pancreatitis widely prevalent in several tropical countries, and fibrocalculous pancreatic diabetes (FCPD) is a form of diabetes secondary to TCP.19 The disease differs from alcoholic pancreatitis by a much younger age of onset, pancreatic calcification, a high incidence of insulin dependent but ketosis resistant diabetes mellitus, and an exceptionally high incidence of pancreatic cancer. Although two studies have reported a lack of common mutations in PRSS1 in FCPD patients, neither of them analysed the whole PRSS1 gene.20,21 The genetic basis of TCP without diabetes has also not been fully investigated. A recent study based on analysis of eight FCPD and four TCP patients (without diabetes) has shown that SPINK1 mutations are associated with FCPD but not with TCP.22 We have analysed 68 clinically and radiologically confirmed TCP patients (24 FCPD and 44 TCP patients without diabetes mellitus) for mutations in the cationic trypsinogen and pancreatic secretory trypsin inhibitor genes in an attempt to understand their respective roles in the pathogenesis of tropical calcific pancreatitis in our population.
SUBJECTS AND METHODS
Patients
Seventy unrelated patients comprising 47 males and 23 females were diagnosed as having tropical calcific pancreatitis based on the following WHO criteria23: (1) recurrent pain in the abdomen since childhood; (2) large intraductal calculi, particularly in the head region; (3) ultrasonological and ERCP evidence of pancreatic calcification; (4) absence of any other aetiological factor like alcoholism, etc; (5) diabetes mellitus as defined by the WHO Study group E (may or may not be present).
DNA analysis
After obtaining informed consent, EDTA blood samples were collected from 68 patients (one male and one female patient refused to participate in the study) and 100 healthy volunteers who served as controls. Genomic DNA was isolated from peripheral blood leucocytes following standard protocols. In view of the extremely high homology of the PRSS1 gene with that of the other pseudogenes, all five exons of PRSS1 were amplified using a nested PCR strategy.9 The primer sequences for the analysis of the promoter region and the four exons of the SPINK1 gene were selected from published sequences.15 The PCR conditions were optimised in a way that the amount of unused primers or unincorporated dNTPs was minimal in the post-PCR reaction mix. All the exons were sequenced individually on both the strands using internal sequencing primers and Big dye terminator cycle sequencing ready kit and an ABI 310 Genetic Analyzer (Applied Biosystems).
Statistical analysis
All values are presented as median (range, 95% CI) and statistical analysis was performed applying the Mann-Whitney U test using SPSSR for Windows software (SPSS Inc, Chicago, IL).
RESULTS
Sequence analysis of the PRSS1 gene
We have analysed all the exons and intron-exon boundaries of the cationic trypsinogen gene in 68 well characterised TCP patients. Sequence analysis of the PRSS1 gene on both strands showed none of the reported mutations like R122H, N29I, A16V, K23R, or D22G nor any novel variant in these patients. This is in contrast to earlier studies, which reported a mutated PRSS1 gene in patients with HP and ICP.6,8,9 Two earlier studies have reported a lack of common mutations (exons 2 and 3 only) in PRSS1 in patients with FCPD and TCP. However, one of them performed a PCR-RFLP assay for the R122H mutation on 11 FCPD and five TCP patients only20 and the other one analysed FCPD patients only.21 In agreement with earlier reports, single C to T transitions in exon 4 (162Asp (GAC-GAT)) and exon 5 (246Asn (AAC-AAT)) without amino acid substitution were identified in the majority of patients (85%) as well as in normal controls (90%).7–9
Sequence analysis of the SPINK1 gene
Direct sequencing of the coding exonic and flanking intronic sequences including the promoter region of SPINK1 detected mutations in 32 out of 68 patients analysed. Of 31 patients with N34S mutation, eight were homozygous (N34S/N34S), 22 heterozygous (N34S/wt), and one compound heterozygous with P55S (N34S/P55S) (fig 1). Only one other patient had a heterozygous P55S mutation (table 1). We have detected SPINK1 mutations in both FCPD patients and TCP patients without diabetes mellitus. This is at variance with a recent study that reported mutated SPINK1 in six out of eight FCPD patients, but none in four TCP patients from Bangladesh.22 We also report the highest frequency of 46% for the N34S mutation reported so far in comparison to 41% (five out of 12 patients) by Rossi et al22 and 26% by Pfutzer et al.16 A novel G to T transversion at 215 bp upstream in the SPINK1 promoter region (−215G>T) was also identified in three patients (fig 2). Interestingly, all three patients also carried an N34S allele, suggesting a compound heterozygote status. Sequencing of 200 control alleles detected three N34S heterozygous mutations (1.5%), one P55S heterozygous mutation (0.5%), but none with the −215G>T mutation. The IVS1-37T>C polymorphism was found to be in complete linkage disequilibrium with the N34S mutation. Another previously reported SNP, −253T>C, was detected in six patients and one of them carried N34S on the other allele. The frequency of this mutation in control chromosomes was 22% with one homozygote and another compound heterozygote with the N34S mutation. This only substantiates the already established notion that −253T>C is a neutral polymorphism.14 No additional mutations were detected in these patients.
Distribution of mutations and polymorphisms in the PSTI gene in patients with tropical calcific pancreatitis and controls
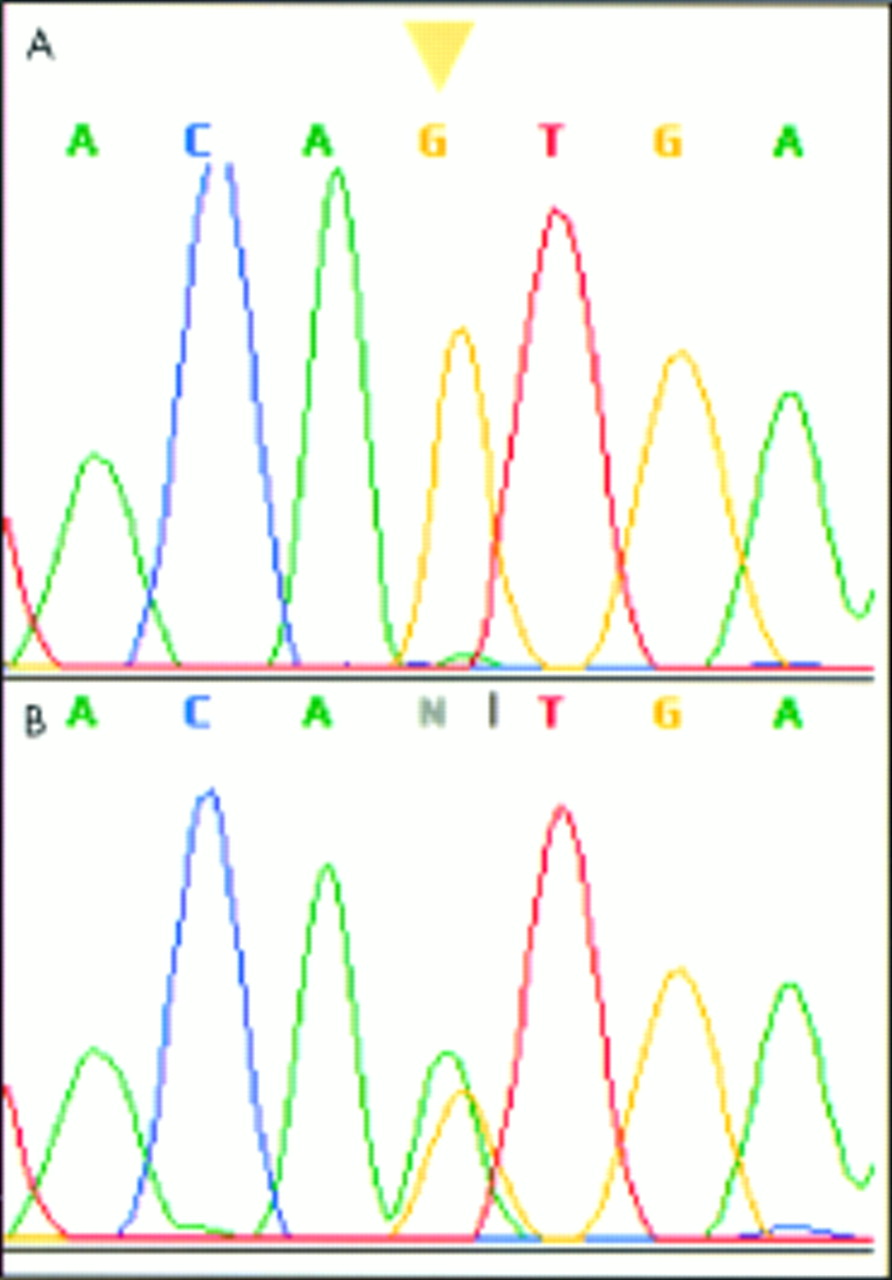
DNA sequencing electropherogram of exon 3 of the human SPINK1 gene showing an A to G transition in a patient with tropical calcific pancreatitis. This mutation leads to an amino acid substitution from Asn to Ser (N34S). The arrowhead indicates the altered nucleotide. (A) Homozygous state. (B) Heterozygous state.

{kind=link}
{kind=link}
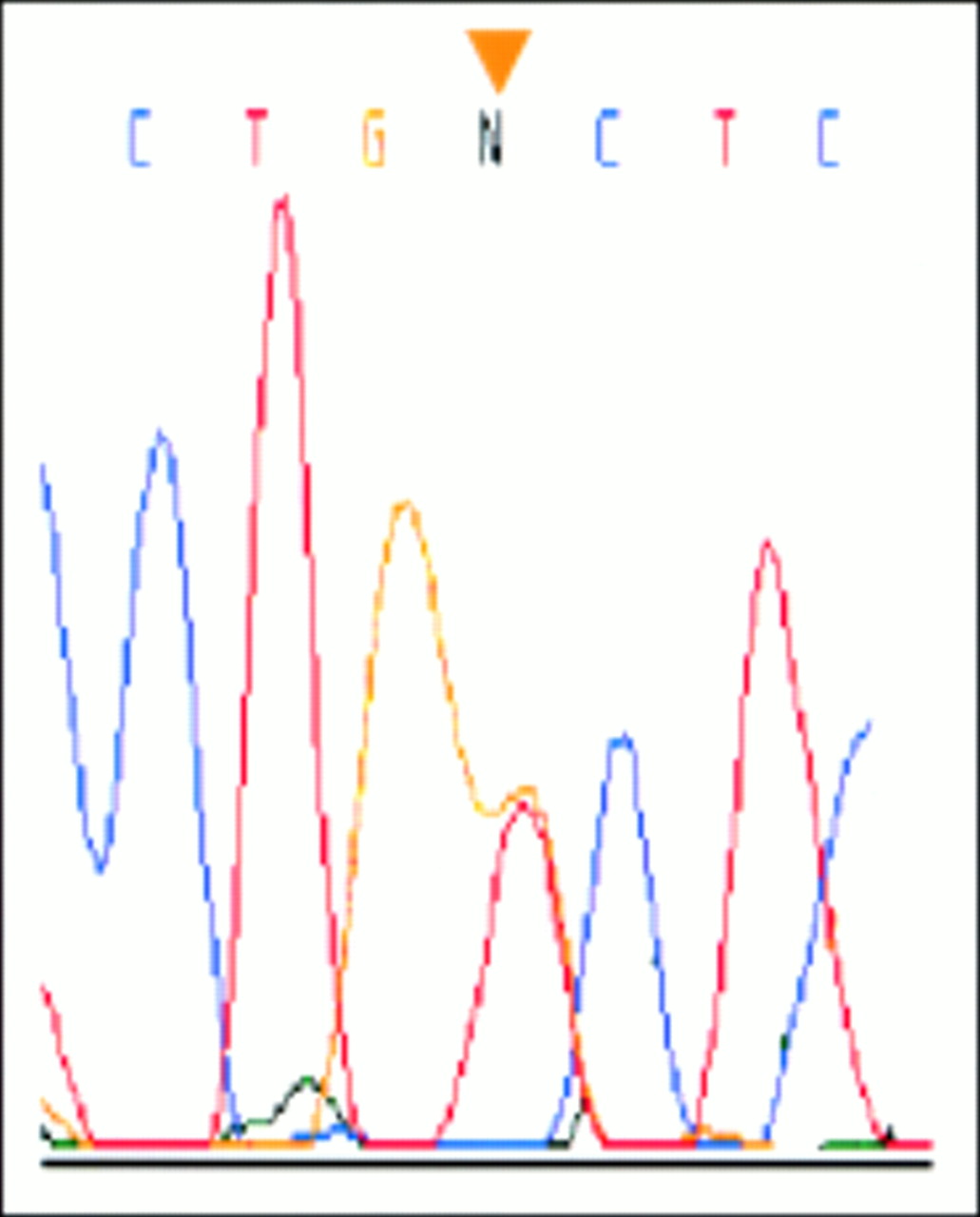
DNA sequencing electropherogram of the promoter region of the human SPINK1 gene showing a heterozygous G to T change at nt 215 upstream from the translation initiation site (−215G>T) in a patient with tropical calcific pancreatitis. The mutation was absent in 200 alleles from normal subjects. The arrowhead indicates the altered nucleotide.
Clinical phenotype
The median age of onset and age of presentation for FCPD patients was 35.0 years (95% CI 27.6-40.2) and 44.0 (95% CI 39.3-49.3), which is significantly higher than that of TCP patients without diabetes mellitus (21.0 years (95% CI 19.9-27.1), p<0.004) and (26.0 (95% CI 25.7-33.0), p<0.001), respectively (table 2). A comparison of SPINK1 mutation frequency in FCPD patients and TCP patients without diabetes showed similar trends, suggesting that neither these mutations nor their status is directly related to the presentation of diabetes mellitus (table 2). No significant difference could be noted on comparison of parameters like the age of onset, presence or absence of diabetes mellitus, and age of onset of diabetes between SPINK1 N34S heterozygous, homozygous, or compound heterozygous patients (data not shown).
Clinical data and status of SPINK1 mutations in the study population
DISCUSSION
Tropical calcific pancreatitis is a variant of chronic pancreatitis of unknown aetiology, occurring mostly in developing countries in tropical regions, and fibrocalculous pancreatic diabetes is a form of diabetes secondary to TCP.24 The aetiology of FCPD is suggested to be multifactorial with both environmental and genetic components.19,25 It has also been suggested that FCPD occurs in a person with susceptibility to diabetes mellitus and might be a distinct entity. The exact relationship between the phenotype of FCPD and TCP is still not clear, although there is an overlap. In our study cohort of 68 TCP patients, 24 were FCPD patients while the remaining 44 were without diabetes at the time of presentation. Comparison of the age of onset shows that FCPD patients had a late age of onset with a difference of more than a decade (21 years v 35 years). It has also been shown that patients with TCP are younger than the FCPD patients and the majority of them have an abnormal glucose tolerance test (GTT).26 This strongly suggests that FCPD may be gradually evolving diabetes in the background of tropical calcific pancreatitis.
In the present study, no mutation could be seen in the cationic trypsinogen gene except two neutral polymorphisms (D162D and N246N) reported earlier.7,9 A pilot study from Bangladesh on 11 FCPD and five chronic calcific pancreatitis patients without diabetes mellitus reported lack of R117H mutation in the PRSS1 gene.20 Subsequent extension of the study to include 50 South Indian and 70 Bangladeshi patients with FCPD excluded common mutations in exons 2 and 3 of the PRSS1 gene.21 Ours is the first report where analysis of the whole coding sequence of the cationic trypsinogen gene does not show any causal mutation in TCP patients. Trypsin plays a central role in pancreatic exocrine physiology by acting as a trigger enzyme leading to activation of all other pancreatic zymogens. A number of protective mechanisms are involved in combating prematurely activated trypsinogen. The current model of PRSS1 mutations suggests that the identified mutations cause enhanced autoactivation of trypsinogen to trypsin or prevent prematurely activated trypsin from being inactivated by autolysis.16 Earlier reports on the presence of identical mutations in Mongolians and the white HP kindreds suggest heterogeneity but no racial specificity.9 Hence, it is surprising that no mutation could be detected in the PRSS1 gene in these patients. Our results show that the established mutations in the cationic trypsinogen gene are not a common cause of tropical calcific pancreatitis in the Indian population.
This study also confirms the causal role of the N34S mutation in the SPINK1 gene in pancreatitis and simultaneously establishes the role of mutated inhibitor in the pathogenesis of tropical calcific pancreatitis. Analysis of the phenotype in terms of the age of onset, frequency of attacks, and presence and age of onset of diabetes mellitus did not show any significant difference between N34S heterozygous or homozygous patients. In addition to the previously reported mutations, we have identified a novel −215G>T mutation which was not found in any of the controls. These three patients were compound heterozygotes with the N34S mutation and had late onset disease with diabetes mellitus, consistent with the FCPD phenotype. This mutation has never been reported previously, although a recent study reported −215G>A in the homozygous state in two out of 32 patients.17 Since SPINK1 N34S heterozygous and homozygous patients have a similar phenotype, it is presumed that mutated SPINK1 cannot cause disease independently by autosomal dominant or recessive mechanisms and the presence of a second mutation, either in the same gene or other genes, is required to express the disease phenotype.16 It may be hypothesised that the mutation −215G>T lies in an unknown cis acting element and may affect transcription of mutated SPINK1 with an already compromised functional capability.17 It may be interesting to dissect the role of this mutation on SPINK1 expression and its interaction with the N34S mutation.
Based on the detection of mutated SPINK1 in FCPD patients only, Rossi et al22 have suggested a genetic basis for FCPD only and also made it the criterion for clinical distinction within TCP with respect to the presence or absence of diabetes mellitus. However, their limited sample size (eight FCPD and four TCP patients) raises serious concern about the validity of such a conclusion. We have detected SPINK1 mutations in both FCPD patients and TCP patients without diabetes (table 2). Our results suggest a common genetic basis for tropical calcific pancreatitis and there could be additional genetic/environmental factors responsible for the variability of phenotype in TCP and FCPD.
Such a high frequency of SPINK1 gene mutations on the background of complete absence of a mutated PRSS1 gene is interesting. To date, no study has reported concurrence of mutated PRSS1 with SPINK1, suggesting that both the mutations work through different and independent mechanisms. However, this pancreatic protease-protease inhibitor system is very important, considering the physiological interaction between them in combatting the prematurely activated trypsinogen inside the pancreas. Intrapancreatic levels of trypsin are expected to be raised if mutations in the inhibitor molecule lead to loss of its inhibitory capacity. However, it is difficult to explain the phenotype of pancreatitis in the presence of normal trypsin when the intact R122 autolysis “self-destruct” mechanism can take care of the prematurely activated trypsin molecule. This suggests impairment of other protective mechanisms involved in combating the prematurely activated trypsin molecule inside the pancreas. The exact mechanism is still not clear and it remains to be proven how the prematurely activated trypsin is sustained inside the pancreatic acini and may cause a low grade inflammation and disease. SPINK1 mutations have been proposed significantly to lower the threshold for pancreatitis from other factors; we suggest the involvement of other genetic/environmental factors in FCPD and TCP patients without diabetes. Several studies have reported mutations in the cystic fibrosis transmembrane regulator (CFTR) gene in both HP and sporadic pancreatitis.27,28 Based on the finding that the vast majority of these patients are heterozygous for the mutation, CFTR has been suggested to act as a modifier gene in the pathogenesis of pancreatitis. A recent study has indicated an association between CFTR mutations and TCP patients; however, only 18 TCP patients have been analysed with two females showing mutations.29 Screening a larger cohort of TCP patients may provide more information about the modifier mutations in the CFTR gene.
In conclusion, our study strongly suggests that cationic trypsinogen gene mutations may not have a role in TCP in our population. We have also shown for the first time the association of mutated inhibitor with tropical calcific pancreatitis, irrespective of the absence or presence of diabetes mellitus (FCPD). The presence of SPINK1 mutations in both FCPD and TCP patients without diabetes mellitus suggests a common genetic basis for tropical calcific pancreatitis. However, different genetic/environmental factors may be involved to account for phenotypic variability in TCP patients. In view of the absence of cationic trypsinogen mutations but significant association of SPINK1 mutations, it may be hypothesised that TCP may occur as a result of mutation in SPINK1 or as a result of a combination of genetic defects of SPINK1 and as yet unidentified genes or environmental factors. As was true for HP acting as a model for understanding the pathophysiology of other types of pancreatitis, the findings from this study will be critical in understanding the variability in the genetic background and association with the variability of phenotype in FCPD and TCP. Considering the fact that about 50% of TCP patients have a mutated SPINK1 gene with the majority having N34S mutation as a founder mutation, patients with pancreatitis may be screened and informed about the risk of developing the disease. An appropriate prevention strategy could be suggested to help the carriers of the mutated allele in modifying the late stage complications of TCP.
-
We investigated 68 patients with tropical calcific pancreatitis comprising 44 without diabetes and 24 with diabetes (FCPD) for mutations in the cationic trypsinogen (PRSS1) and pancreatic secretory trypsin inhibitor (SPINK1) genes. We also compared the clinical phenotype of FCPD and TCP without diabetes in terms of age of onset, severity of symptoms, frequency of mutated SPINK1, and its association with the two phenotypes.
-
Our study could not detect any mutation in the PRSS1 gene, strongly suggesting that cationic trypsinogen gene mutations may not have a role in TCP in our population.
-
We detected mutated SPINK1 in about half the patients with a founder mutation N34S in the majority of the patients. We also report a novel mutation −215G>T in compound heterozygosity with N34S in three FCPD patients. In contrast to a recent report, we identified SPINK1 mutations in both FCPD and TCP patients without diabetes at a similar frequency.
-
The median age of onset for TCP patients without diabetes mellitus (21.0 years (95% CI 19.9-27.1 )) is more than a decade earlier than that of FCPD patients (35.0 years (95% CI 27.6-40.2), p<0.004). Comparison of parameters like the age of onset, presence or absence of diabetes mellitus, and age of onset of diabetes between SPINK1 N34S heterozygous and homozygous patients did not show any significant difference.
-
These data suggest a common genetic basis for tropical calcific pancreatitis with additional genetic/environmental factors responsible for the variability of phenotype in FCPD and TCP patients without diabetes.
Acknowledgments
GRC expresses deep gratitude to Dr Ranvir Singh, CCMB, for helpful discussions. Our thanks are also due to all the patients, their families, and people who voluntarily participated in the study. Financial support from the Indian Council of Medical Research, New Delhi is also acknowledged.