Article Text

Statistics from Altmetric.com
Hereditary non-polyposis colorectal cancer (HNPCC) syndrome is a dominantly inherited condition, recognised by Lynch et al1 in 1966, associated with a major susceptibility to colorectal and endometrial cancer. The use of stringent criteria in the definition of the syndrome led to the initial identification of two genes, MSH2 and MLH1, which when mutated are responsible for the cancer susceptibility. Mutations in these two genes may account for up to two-thirds of all HNPCC cases.2 More recently, additional genes such as MSH6, PMS1, PMS2, MLH3, TGFβRII, and EXO1 have also been implicated. Together deleterious mutations of these genes account for less than 10% of the cases.
In addition to the colorectum and endometrium, a variety of different sites have been reported to be at an increased risk of cancer in HNPCC patients. Indeed, studies based on either family history or on small groups of patients with identified mutations suggested that carcinomas of the ureter, stomach, small bowel, ovary, and biliary tract1,3 were more frequently encountered than in the general population. In addition to this tumour spectrum, the presence of sebaceous skin tumours and keratoacanthomas or of glioblastomas defines two different subsets within the HNPCC syndrome, called Muir-Torre4 or Turcot5 syndromes, respectively. Finally, more recently, an excess of breast cancer has also been reported but remains controversial.6
Following these descriptions, several management and follow up programmes for HNPCC families have been proposed.7,8 To delineate these procedures more precisely, it would be useful to gain further insight into the associated risks of MSH2 and MLH1 mutations.
Prospective recruitment of 436 consecutive patients referred to a French family cancer clinic enabled the identification of 348 subjects from 163 families with an established deleterious mutation in either MSH2 or MLH1. Analysis of the tumour history of these subjects enables a critical review of some of the presently proposed procedures for their management.
PATIENTS AND METHODS
All consecutive probands referred to the family cancer clinic between January 1995 and November 2000 and satisfying at least one of the modified Amsterdam criteria9 were recruited to participate in the study. Patients with a deleterious mutation were asked to contact their at risk relatives. A total of 320 at risk relatives presented at the clinic and all agreed to take part in the study and provided written informed consent. Gene carriers were followed prospectively after their inclusion.
The clinical data were collected by SO through patient interviews and hospital notes. Sex, date of birth, date of death or last medical interview, and type of germline mutation were systematically recorded for each gene carrier. For those having developed tumour(s), age at diagnosis and location was recorded for each cancer. Whenever possible, histological confirmation was collected from pathological reports.
Genetic testing
Blood samples were obtained from all participating subjects and kept frozen at −30°C until DNA extraction.
Key points
-
MSH2 and MLH1 mutation carriers are susceptible to a wide spectrum of cancers, predominantly in the colorectum and endometrium. To improve identification and management of these patients, age at onset and location of each cancer that had affected a series of 348 HNPCC gene carriers were recorded.
-
Tumour histories of MSH2 and MLH1 patients were remarkably similar. Ages at first tumour were not correlated within families. Tumours that developed before 30 years were all located in the colon or rectum. Five percent of gene carriers experienced cancer before 25 years of age. Colorectal cancer risks for both sexes were comparable but delayed in females by 5 to 10 years. Colorectal cancer was the first manifestation of the disease in 89% of affected males but in only 66% of affected females. Endometrial cancer was the first manifestation in 26% of affected females.
-
Subjects with colorectal or endometrial cancer aged less than 50 should be invited for screening for MSH2 and MLH1 mutation. Surveillance of gene carriers should be initiated in early adulthood, that is, 18 years, and should not be delayed by the age at onset of the index case. Up to the age of 30, surveillance should focus on the colorectum for both sexes.
Systematic screening for point mutations in the MSH2 and MLH1 genes was performed on DNA from each independent proband using denaturing gradient gel electrophoresis (DGGE). Analyses of the sequence of the MSH2 and MLH1 genes (reference sequences NM_000251 and NM_000249) were performed by specific computer programs (Melt87 and SQHTX) in order to optimise the selection of pairs of amplimers and the electrophoretic conditions that would enable detection of single base pair substitution anywhere in the coding sequences of the MSH2 and MLH1 genes or in their adjacent splicing consensus sites. Because of AT rich domains, exons 2 and 5 of MSH2 and exon 12 of MLH1 could not be screened reliably by this method and were directly sequenced after PCR amplification.
The MSH2 and MLH1 genes were screened for genomic rearrangements using a quantitative multiplex PCR approach, which has been previously described.10 De novo mutations were ascertained by typing eight microsatellite polymorphisms in the DNA of the patient and living parents.
Statistical analyses
Age specific cumulative incidence rate for first and second tumours according to sex, mutated gene, and localisation were analysed with the log rank test. Anova was used to test for intrafamilial correlation of the age at onset of the first tumour.
RESULTS
Gene carrier recruitment
A total of 436 probands referred to the family cancer clinic between January 1995 and November 2000 satisfied one of the following criteria: group A (245 cases) fulfilled all three modified Amsterdam criteria9; group B (71 cases) included affected patients with one affected first degree relative, at least one of the family members (proband or relative) being affected younger than 50 years; group C included sporadic cases with colon cancer diagnosed under 50 years of age. This group was further subdivided into a subgroup of patients who had a second cancer at the time of the recruitment (group C1, 41 cases) and those who did not (group C2, 79 cases). Subjects with multiple primary tumours and at least one affected first degree relative were classified into either group A or B according to their family history. Corresponding DNA was screened to search for mutation in MSH2 and MLH1 (see below) resulting in the identification of a deleterious mutation in 163 probands (15-64 years, mean age 40.1). Subjects with sporadic cancer before the age of 50 were shown to be gene carriers in 22% of the cases. As expected, this proportion was higher for multiply affected cancer patients (14 of 41 screened subjects, 34%) as compared to singly affected patients (13 of 79, 16%).
Probands with a MLH1 or MSH2 mutation were asked to contact their relatives about the possibility of genetic testing and subsequent management for gene carriers. A total of 320 relatives from 67 families came to the clinic and 153 were found to be gene carriers. At that time, genealogical data allowed the inclusion of 32 additional obligate carriers under the assumption that the deleterious mutations observed in the members of a single kindred were identical by descent. Among the 185 gene carriers, 100 were asymptomatic (18-71 years, mean age 32.8) and the disease had already affected 85 (20-85 years, mean age 41.5). Thus, at the end of the recruitment procedure, a total of 348 gene carriers from whom clinical data could be collected were identified. In addition, 167 non-gene carrier first degree relatives of a gene carrier were identified.
Spectrum of DNA variations in MSH2 and MLH1 patients
All exons and adjacent intronic regions of MSH2 and MLH1 were screened for all 436 probands (for DGGE conditions, see electronic supplementary table 1), leading to the identification of 167 different DNA variants. Of these, 24 silent or intronic variants located outside the consensus splicing sites were not considered further.
Distribution of tumours according to sex of patients and mutated gene
Among the 143 remaining variants, 84 were predicted to alter the open reading frame (36 nonsense mutations, 29 frameshift mutations, and 19 mutations in consensus splicing sites); such mutations were found in 108 index cases (electronic supplementary table 2). They were readily classified deleterious. The rest consisted of five small in frame deletions and 49 missense mutations (electronic supplementary table 3). In order to document their potential implication in the disease, six pieces of information were collected (electronic supplementary table 3). Twenty missense variants were excluded because they met none of these criteria. They have been described elsewhere as non-functional and several of them have been reported as frequent polymorphisms (MSH2: Q10K, V78A, P259S, G322D, L556V, T557P, R621Q, E853G, I884S; MLH1: A31C, V213M, I219V, Q346H, G454R, Q562P, K618T, P640L, A681T, H718Y, H733Y).14,15,17,18 Two mutations showed only the phylogenetic conservation criteria. One of them, a three amino acid deletion (position 742-744 of MLH1) was classified as deleterious because it occurred in the highly conserved CTH motif, which when deleted had been shown to impair MLH1 function.14 The second, L330P of MSH2, was predicted to play an important role in the interdomain interactions.11 Further somatic characterisation of tumour cells showed a high level of microsatellite instability using the international panel of markers,7,8 and an absence of MSH2 immunoreactivity using the NA27 FE11 monoclonal antibody on formalin fixed tissue sections (Calbiochem Oncogene Research Products, data not shown), so it was also retained as deleterious. Two mutations were predicted to abolish normal translation by removing the initiation codon. The remaining 30 mutations were predicted by at least two criteria to alter the normal function of either protein. Thus, in total, 34 different missense or small in frame variants were classified as deleterious. They were found in 51 different index cases.
Finally, a search for genomic rearrangements enabled four large deletions to be identified, all involving the MSH2 gene (deletions of exon 5, exons 5 and 6, exons 9 and 10, exons 1 to 15, respectively).10
Thus the presence of a deleterious MSH2 or MLH1 mutation could be documented in a total of 163 index patients.
Clinical history
The clinical history of these 163 patients and their 185 gene carrier relatives was further investigated by examination of their medical files and by patient interviews.
Distribution of tumour location
The distribution of the tumour location appeared very similar when patients were classified according to their deleterious gene or to their mode of recruitment (that is index cases v family relatives). Approximately half of the tumours were colon carcinomas located proximal to the splenic flexure. In males, a quarter of the tumours were distal colonic or rectal cancers. In females the proportion at this location was smaller (19%) and endometrial cancer accounted for a quarter of the diagnosed tumours. The small intestine accounted for 6% of the tumour location in index cases. It was less than 1% in the family relatives (Fisher exact test p<0.01). The rarer locations (stomach, skin, central nervous system, ovary, biliary tract, kidney, and urinary tract) accounted collectively for 10% of the locations (table 1). Of interest was the fact that breast, thyroid, lung, and prostate cancer were infrequent for both groups of patients suggesting that patients with a deleterious mutation in MSH2 and MLH1 may not be at increased risk for these cancers.
Frequency of tumour and age at onset
Ages at tumour onset of MSH2 and MLH1 index cases were similar (40 ± 9 years for both genes). It was about five years older for gene carrier relatives (MSH2 46 ± 12, MLH1 44 ± 12). Within both groups, the age specific cumulative incidence rates were compared using time series analysis taking censoring into account. They were not found to be significantly different when patients were classified according to the mutated gene (Wilcoxon test, p>0.3; in both cases, a shift of five years would have been detected at the level of 0.05 with a power close to 1). The age at which half the index cases became symptomatic was 40 and was 48 for family relatives. This difference may be explained to a large extent by the different recruitment modes of the two groups of patients. Before their recruitment, index cases had to be symptomatic and therefore the evaluation of the age dependent cumulative risk of becoming symptomatic in this group of patients is overestimated. Conversely, with the exception of 32 obligate carriers who did not provide blood samples, all 153 family relative carriers had to be alive at the time of DNA testing, thus inducing a bias towards underestimation of the risk of being symptomatic.
A risk estimate for becoming symptomatic for a given age group, called Rs, that is less dependent on the recruitment mode of index cases and families, can be obtained by counting, among unaffected offspring of gene carriers in this age group, the number of asymptomatic gene carriers Nas and the number of non-gene carriers Nng. These three quantities are related as:
This approach was applied to two age groups. In the age group between 30 and 45, Nas and Nng were found equal to 31 and 55. The mean and variance of the age distributions were similar (38 ± 5 years). Thus, at 38, the risk of becoming symptomatic is estimated to be close to 0.43. In the age group 45-60, Nas and Nng were equal to 11 and 29 respectively. The average age was 51, again not being significantly different in the two groups. The risk of becoming symptomatic should be close to 0.62 at that age (fig 1). The age specific cumulative incidence rate for various tumour locations in index and family relatives may represent the upper and lower estimates for gene carriers. Because evaluation of the risk of tumour development may be more accurate if data from index and family relatives are merged, such integration was performed in the subsequent analyses.

Age specific cumulative incidence rate of cancer. Continuous and dotted lines represent the risk of index cases and gene carrier relatives respectively to develop a cancer of the HNPCC tumour spectrum. Merged data are displayed on the thick curve. The two stars indicate the risk evaluation calculated from asymptomatic offspring of gene carriers (see text).
In the present series, variance analysis of the age at onset of the first tumour in affected subjects failed to show a significant difference among families (p=0.26; simulation showed that a coefficient of intrafamilial correlation of 0.5 would have been detected with a power of 0.94 at a significance level of 0.05). Mutations were classified into two groups according to their structural consequence. Group 1 included all missense mutations leading to a single amino acid change or a single amino acid deletion. Group 2 included those mutations predicted to cause a gross alteration of the protein structure (protein truncation or large interstitial deletion in the polypeptide sequence). Such alterations could be promoted by large genomic deletions, frameshift mutations, nonsense mutations, splice site mutations, and deletions predicted to cause loss of several amino acids. Gene carriers were classified according to their mutation being in groups 1 and 2; the age specific cumulative incidence rates of first tumour were compared in both groups and were not found to be significantly different (Wilcoxon test, p=0.1, a difference in five years would have been detected with a power close to 1 at a significance level of 0.05) (fig 2). Taken together, these observations suggest that, regardless of their type, most or all mutations studied here have similar consequences (that is, inactivation of the mutated allele), an observation in accord with the absence of strong intrafamilial correlation.

Age specific cumulative incidence rate of cancer in gene carriers. Graph A represents the cancer risks according to the mutation consequence. Mutations of group 1 are shown by the dotted line and of group 2 by the continuous line. Graph B shows the risk of endometrial cancer according to the mutated gene (MSH2 on continuous line, MLH1 on dotted line). Graphs C and D represent the risks of proximal and distal or rectal cancer respectively, according to the sex of the gene carriers (continuous line for males, dotted line for females).
When Kaplan-Meyer plots were performed for proximal colonic cancer or for distal colonic and rectal cancer, classifying patients according to the nature of the deleterious gene (MSH2 v MLH1), no significant difference was observed. Although endometrial cancer appeared more frequent in MSH2 female patients, analysis of the Kaplan-Meyer plots failed to show statistical significance (Wilcoxon test, p=0.24). We conclude that, in our series, deleterious mutations in MSH2 and MLH1 have very similar clinical consequences. Interestingly, on average, colorectal cancer appeared earlier in males than in females. This difference was true both for proximal colonic tumours (Wilcoxon test, p=0.002) and for distal colonic and rectal tumours (Wilcoxon test, p=0.00004) (fig 2). In the latter case, on average males would develop a cancer at that location about 10 years earlier than females. Importantly 5% of the gene carriers (14 cases) had developed colorectal cancer before 25 years of age. The frequency of small intestinal tumours appeared more frequent in males than in females (Fisher exact test, p=0.009). Strikingly, although infrequent, all observed ovarian cancer developed between 35 and 44 years of age, that is, at premenopausal age.
Location of first tumour at disease onset
Although the age at onset was not significantly different between males and females, the location of first tumour differed between the two groups. A proximal colon cancer was the first neoplasm to develop in 85 of 126 symptomatic male gene carriers (67%) and 63 of 122 symptomatic females (50%). The risk for distal and rectal cancer being the first tumour was substantial, being 22% (27 of 126) and 14% (17 of 122), respectively. Thus, colorectal cancer being the first tumour was observed in 89% of affected males but in only 66% of affected females. Endometrial cancer was the first and only tumour at disease onset for 26% of the affected female gene carriers. All tumours that occurred before 27 years of age were located in the colon or rectum. Earliest non-colorectal cancers being the first tumour were a small intestinal cancer diagnosed at 27 years, a gastric cancer diagnosed at 30, and an endometrial cancer diagnosed at 30. Tumours of the small intestine were the first and only tumour at disease onset on seven occasions.
Risk of metachronous tumour
Multiple cancers affecting the same patient were observed for 91 mutation carriers (36% of the symptomatic cases). This proportion was similar for index cases (63 of 163, 39%) as compared to their affected family relatives (28 of 87, 32%). The risk for a second tumour was similar for MSH2 and MLH1 patients, as well as for males and females.
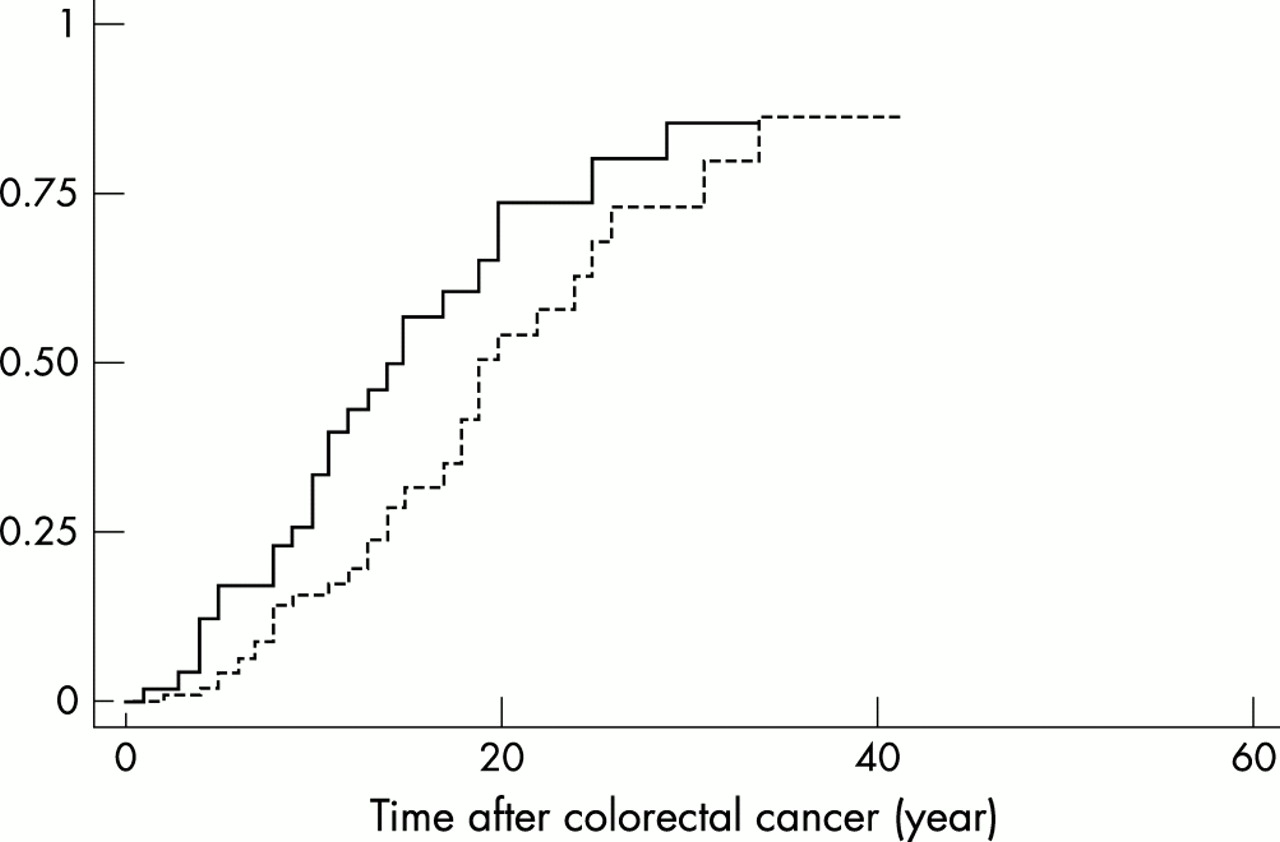
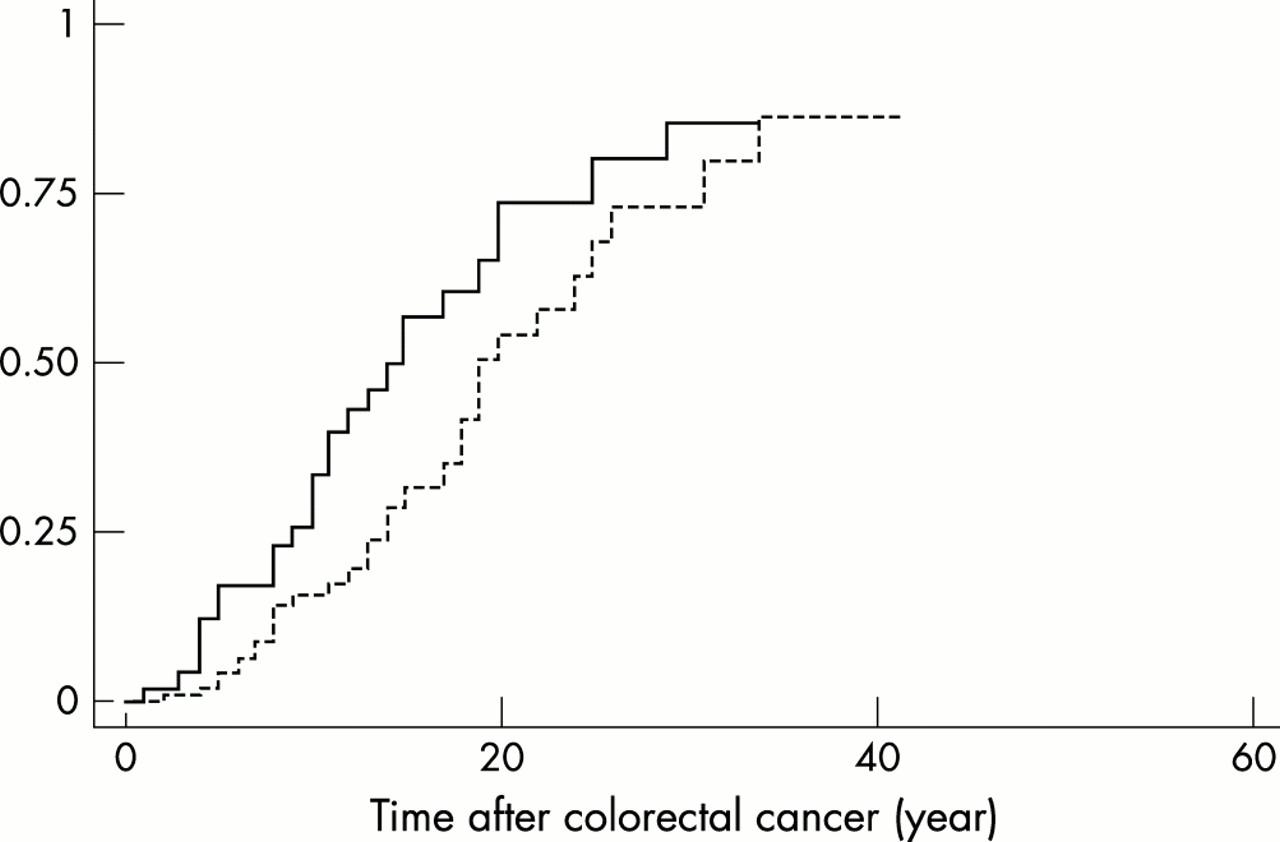
The age specific cumulative incidence rate for developing a colon cancer for two different localisations of the first tumour was investigated. The first localisation to be considered was the colon or rectum. Because at the time of surgery no patient had been diagnosed as a gene carrier, segmental colectomy had been preferentially performed. Thus, the remaining portion of the colon and/or rectum could subsequently be the site of a metachronous cancer. This happened on 34 occasions. The second localisation of the first tumour to be considered was the uterus. In spite of the observation that females are at lower risk of developing colorectal cancer, the development of a second cancer of the colon or rectum occurred on average earlier in this group as compared to the previous one (fig 3). This observation may have two non-exclusive explanations. First the target site for the second cancer was smaller in the first group of patients who had undergone a segmental colectomy; second, in this group, patients were under colonoscopic surveillance so that adenomas could be resected before their progression to cancer. In contrast, because patients with endometrial cancer had rarely been identified as HNPCC patients, their large bowel was not under surveillance.
{kind=link}
{kind=link}
{kind=link}
Risk of metachronous colorectal cancer. The age specific cumulative incidence rate of colorectal cancer is plotted according to the previous cancer, being endometrial for the continuous line and colorectal for the dotted line.
DISCUSSION
The present study reports a series of 163 unrelated patients affected with colorectal cancer, for whom a deleterious mutation in either the MSH2 or MLH1 gene could be determined, and the subsequent identification of 185 gene carrier relatives. Mutations were observed in all exons indicating that exhaustive screening for mutation in newly recorded cases requires analysis of the entire coding sequence. Furthermore, it has been recognised that MSH2 and MLH1 are the sites of various large genomic deletions or insertions,19 indicating that these alterations have to be searched for to ensure a more complete investigation.
It has been previously reported that MSH2 mutation carriers tend to be at a higher risk of cancer compared to MLH1 mutation carriers.6 In the present set of data, a similar trend has only been observed for endometrial cancer. However, it may be small as the difference did not reach statistical significance. More generally, analysis of the present series failed to detect statistically significant differences between patients carrying mutations in either the MSH2 or MLH1 gene for any tumour site. This observation indicates that alterations in MSH2 or MLH1 may have similar consequences on DNA stability.
The mode of inactivation of MSH2 and MLH1 in HNPCC patients appears comparable. For both genes, the relative proportion of the group of missense mutations and small deletions compared to that of truncating and large deletions is equivalent. The observation that the age dependent risk of tumours is similar for both types of mutations indicates that most or all missense mutants classified as deleterious in this work have failed to retain a significant residual function or may not play a dominant negative role, which would have facilitated tumour initiation. At variance with familial adenomatous polyposis where the position of mutation along the APC gene has been associated with different clinical outcomes,20–22 in HNPCC patients most MSH2 and MLH1 mutations may have very similar clinical consequences. Thus, a unified procedure may be proposed for the management of most MSH2 and MLH1 HNPCC patients.
A precise knowledge of the age dependent risk of cancer for subjects with deleterious MSH2 and MLH1 mutations is necessary for their optimal follow up. Such evaluation, when performed on symptomatic subjects referred to a family cancer clinic, is biased by the mode of recruitment. To reduce the consequence of this ascertainment bias, we have proposed here an evaluation based on the age dependent proportion of gene carriers observed in asymptomatic offspring of gene carriers. This method is unbiased by the mode of recruitment of the affected subjects and families. However, in order to obtain good precision using this method, a large number of asymptomatic offspring of gene carriers, distributed over an extended range of age, is required. The use of this method on the present recruitment has led to a first evaluation of the age dependent risks of first cancer to be approximately 0.43 and 0.62 at ages 38 and 51, respectively. Larger series will have to be used to improve the precision of these estimates. Kaplan-Meier plot of the pooled data from index cases and family relatives evaluates the ages at which these risks are reached as 40 and 48 years, respectively, suggesting that such pooling may provide for the present series an approximation of the age specific cumulative incidence rate of first cancer.
The present risk evaluations are larger than those inferred from the study of Vasen et al,6 which is based on the analysis of 74 extended families with MLH1 or MSH2 mutation or from that of Dunlop et al,23 based on the analysis of six extended families recruited from a single case of colorectal cancer before the age of 35. The discrepancies in the three studies may in part be because of the recruitment methods. However, differences in endometrial cancer risk and age at onset of the first tumour between Korean and Dutch HNPCC families has recently suggested that environmental or perhaps modifier genes may significantly modulate the cancer risk.24 In the absence of further insight into the causes of the discrepancies between the series, the present risk evaluation may be used as a base to discuss the optimal management of the HNPCC syndrome in France.
Strikingly, all 21 tumours that developed before 30 years of age affected the digestive tract. Two small intestinal tumours developed at 20 (in a patient who had a distal colon tumour at 18) and 27. All other tumours were located in the colorectum, suggesting that colonoscopy alone should be an effective surveillance method. Although distal colonic and rectal cancer are delayed by 10 years in gene carrier females as compared to males, the delay for proximal colon cancer is much smaller. It may therefore not be advisable to delay colonoscopy in female gene carriers. These sex differences may not be specific for HNPCC, since a similar age difference has been noted for sporadic colorectal cancer.25
The fact that endometrial cancer has never been reported before the age of 303,6,23,26,27 (present work) shows that uterine surveillance may be delayed until this age. In fact, apart from the colorectum and small intestine, all tumours at all other sites developed after 30. In contrast, 5% of the patients developed a colorectal tumour before 25 years of age, suggesting that periodic colonoscopy initiated in adulthood may be an option for early follow up. Such surveillance remains to be evaluated and the optimal frequency of colonoscopy should be determined. After the age of 30 multiple sites have been proposed to be regularly monitored, but efficiency and/or acceptance were not assessed.29,30 It has been intended that the surgical treatment of any colon carcinoma in HNPCC patients is total colectomy. Given the risk of metachronous colonic or rectal cancer (fig 3) and the technical difficulty in performing a total colectomy after a segmental resection, this therapy appears relevant. The risk of a second colorectal cancer was comparable when the first cancer occurred in the colon and in the uterus. However, in this case, it appears more advisable, given the success of screening programmes,28 to propose regular colonoscopy to the patients instead of preventive total colectomy.
In conclusion, the absence of significant genotype-phenotype correlation justifies the clinical evaluation of a unique surveillance protocol in the case of MSH2 or MLH1 germline mutations that might be initiated from the start of adulthood for colorectal cancer risk and at 30 years for other sites. Finally, because endometrial cancer is the first presenting symptom for a quarter of gene carrier females, a personal or familial history compatible with HNPCC disease should be systematically investigated.
Acknowledgments
We thank the Ligue Nationale Contre le Cancer (comité aubois), Association pour la Recherche sur le Cancer, and Société Nationale Française de Gastro-Entérologie (fonds de recherche) for financial support. HNPCC-France and all the French clinicians, who referred HNPCC patients to our institution, particularly the FNCLCC network (Fédération Nationale des Centres de Lutte contre le Cancer) are gratefully acknowledged.
REFERENCES
Supplementary materials
Cancer risk in 348 French MSH2 or MLH1 gene carriers
Y Parc, C Boisson, G Thomas, and S OlschwangWeb-only Supplementary Tables
Table 1 PCR amplification and DGGE conditions
[View PDF]
Table 2 MSH2 and MLH1 truncating mutations
[View PDF]
Table 3 Missense variants with predicted biological consequence
[View PDF]