Article Text

Statistics from Altmetric.com
- CT, computed tomography
- ELISA, enzyme linked immunosorbent assay
- ERCP, endoscopic retrograde cholangiopancreatographic examination
- HP, hereditary pancreatitis
- PSTI, pancreatic secretory trypsin inhibitor
- SPINK1, serine protease inhibitor Kazal type 1
Hereditary pancreatitis (HP) is an inborn disorder which leads to recurrent episodes of pancreatitis in children and young adults and is associated with exocrine pancreatic insufficiency and secondary diabetes.1–3 Several germline mutations in the cationic trypsinogen (PRSS1) gene have been found to be associated with the disease phenotype, the most common of which are the R122H, N29I, and A16V mutations.4–6 The clinical significance of various other PRSS1 mutations that have been reported from isolated patients or single families is as yet unclear.7,8 In many families with HP some relatives who carry the mutation relevant to the disease will never develop pancreatitis. The prevalence of these healthy carriers varies from between 20% and 30% in families with the common R122H and N29I mutations,2,5 and may rise to nearly 80% in families with the A16V mutation.6,9 The underlying cause for this variable penetrance remains unknown, but environmental as well as genetic factors might be involved.
Key points
-
Hereditary pancreatitis (HP) is strongly associated with mutations in the cationic trypsinogen (PRSS1) gene but the phenotypic penetrance varies between 20% and 80%. Why some carriers of mutations never develop the disease phenotype is unknown.
-
In 100 patients and relatives from 40 unrelated HP kindreds with a PRSS1 mutation, we have investigated whether coinherited mutations in the serine protease inhibitor, Kazal type 1 (SPINK1) gene, affect the onset, penetrance, and clinical severity of pancreatitis.
-
Among 17 unaffected carriers of PRSS1 mutations and 70 patients with pancreatitis with a PRSS1 mutation (11 with diabetes), we found the following SPINK1 alterations distributed across six (15%) families: N34S mutations (7), 3′UTR 272 C>T polymorphisms (3), and 3′ flanking region 371 G>A polymorphisms (1). Carriers of N34S were as frequent among patients with and without diabetes (9% v 7%), had no earlier disease onset (median age 12 v 10 years), and were found among healthy PRSS1 carriers (12%) and patients with pancreatitis (6%). There was no correlation between the age at onset of disease and either disease severity or prevalence of diabetes.
-
SPINK1 mutations, which play a part in the pathogenesis of idiopathic pancreatitis, are relatively uncommon among patients with HP with a PRSS1 mutation and, when present, affect neither the disease phenotype nor its penetrance. Patients in whom pancreatitis begins in childhood are no more at risk of developing severe disease or diabetes than patients who develop pancreatitis as adults.
More recently, mutations in the gene encoding the most abundant physiological inhibitor of digestive proteases, the serine protease inhibitor Kazal type 1 (SPINK1), also referred to as pancreatic secretory trypsin inhibitor (PSTI), have been found to be associated with idiopathic chronic pancreatitis.10,11 Although the biochemical role of SPINK1 in the onset of disease remains to be elucidated, SPINK1 mutations are clearly involved in the pathogenesis of different varieties of pancreatitis12–14 including alcohol induced pancreatitis.15 One possible explanation for this association is that trypsin seems to be critically involved in the premature activation of other digestive proteases16,17 and SPINK1 is its most potent endogenous inhibitor. Structural alterations in SPINK1 could thus affect the protease/antiprotease balance within the pancreas and this may be of particular importance in HP where the premature activation of digestive proteases is thought to play a predominant role.4,16
We have tested this hypothesis and studied whether SPINK1 mutations (1) segregate with known HP relevant trypsin mutations, (2) if present, aggravate the disease phenotype or increase the prevalence of secondary diabetes, and (3) determine the disease penetrance of HP. We also investigated whether onset of disease in early childhood would aggravate the severity of the clinical course of the disease or prevalence of diabetes.
METHODS
The study population consisted of patients and relatives from 40 unrelated kindreds with HP who had been referred to two tertiary referral centres in Münster and Berlin. Only families with documented disease relevant PRSS1 mutations were included. The prevalent PRSS1 mutations in those kindreds were R122H (25), A16V (10), N29I (3), R122C (1), and R116C (1). Patients and relatives were offered genetic research testing for SPINK1 mutations according to established guidelines and ethics committee regulations.18 From the 100 subjects who consented to participate in the study, 17 were healthy carriers of PRSS1 mutations and 70 were patients with pancreatitis (11 with diabetes). For 21 families, complete clinical and genotype data for PRSS1 and SPINK1 across two or more generations were available. Clinical data were obtained by structured interviews with patients and, particularly in the case of children, with relatives, as well as from hospital charts. Patients were specifically asked about their age at onset of disease, pancreatic surgery, history of endoscopic interventions, and the presence and onset of insulin dependent diabetes mellitus. In the final analysis disease severity was classified as either mild or severe. Mild disease was assumed when no gross structural abnormalities of the pancreas were found on endoscopic retrograde cholangiopancreatographic examination (ERCP) or computed tomography (CT) scans, and the patient had no exocrine pancreatic insufficiency and no history of pancreatic surgery. Severe disease was assumed when patients had undergone pancreatic surgery, had exocrine pancreatic insufficiency, or had structural changes of the pancreas on imaging studies classified as stage III or greater according to the Cambridge classification.19 Exocrine pancreatic insufficiency was assumed when faecal elastase concentration was <200 μg/g in two separate measurements (enzyme linked immunosorbent assay (ELISA), ScheBo Tech, Wettenberg, Germany) and the patient clinically required oral pancreatic enzyme replacement.
A group of 10 patients who were classified as having moderate disease because they fulfilled characteristics of the mild as well as the severe disease group were omitted from the final severity analysis, although their data on disease onset were not significantly different from those of both other groups. In these few patients either severe changes on imaging studies were not paralleled by exocrine pancreatic insufficiency or, alternatively, overt exocrine pancreatic insufficiency was not associated with correspondingly severe morphological changes.
Leucocyte DNA was extracted from EDTA treated blood samples with spin columns (Qiagen, Hilden, Germany). The complete SPINK1 coding region was amplified by polymerase chain reaction with specific primers (PSTI-S-1 aaccagggagatctgtgata, PSTI-AS-1 gtcagccacatcaatagagg, PSTI-S-2 ggggtacaggaaaatcagtc, PSTI-AS-2 ccttcacagcaagcactgta, PSTI-S-3 agaaatagcagaggcatgac, PSTI-AS-3 ggcttttatcatacaagtgac, PSTI-S-4 ctcaaacctctccaacttta, and PSTI-AS-4 gagggaaaccctgtctgaaa) or as already described.10 Direct DNA sequencing was performed with the BigDye terminator mix (Applied Biosystems) and an ABI Sequencer. The t test, χ2 test, and Pearson and Spearman correlation coefficients were used for statistical evaluation and differences of >5% were considered significant. Graphs include cumulative incidences, regressions, and medians with 90th centiles and error bars.
RESULTS AND DISCUSSION
To answer the question of phenotypic penetrance, sufficient families that include unaffected carriers of a disease relevant mutation needed to be studied. We recruited 17 healthy carriers of PRSS1 mutations from a total of 13 HP kindreds and sequenced their SPINK1 gene. Three of these 17 PRSS1 carriers (18%) were found to also carry genetic alterations of the SPINK1 gene (N34S mutations (2), 3′ flanking region 371 G>A polymorphism (1)). Among patients with pancreatitis eight (11%) carried a SPINK1 variation (N34S (5), 3′UTR 272 C>T (3)). This distribution alone makes a role of SPINK1 in the onset of HP associated with PRSS1 highly unlikely. The most persuasive argument, however, against a determining role of SPINK1 in the penetrance of HP comes from kindreds in whom relatives with a trypsin mutation as well as the disease relevant N34S SPINK1 mutation were found among patients with pancreatitis and healthy carriers alike (fig 1A, B). In other extended kindreds, patients with pancreatitis and carriers with and without N34S mutation were randomly distributed (fig 1C).
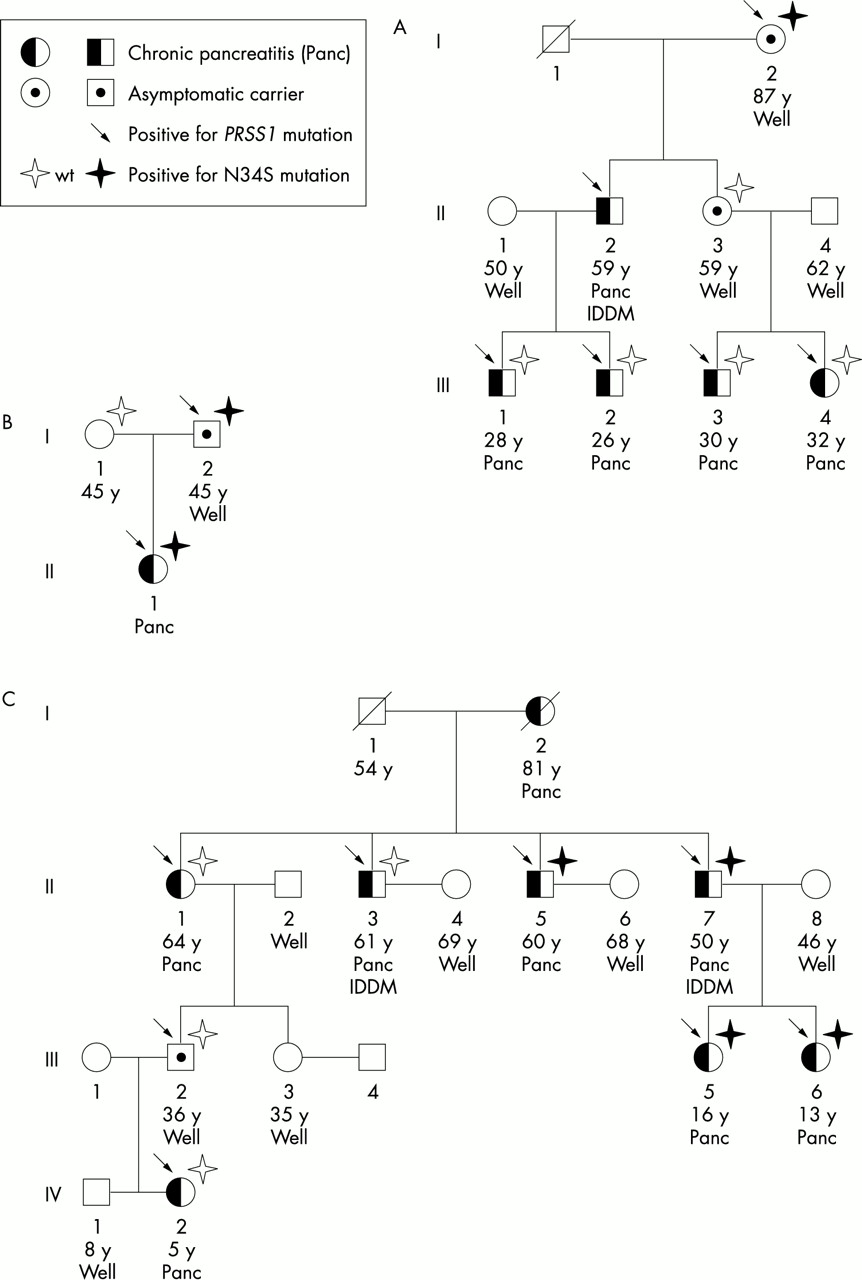
Three informative pedigrees of families with HP and coinheritance of the N34S SPINK1 mutation. In family A (R112H trypsinogen mutation), carriers with and without the N34S mutation were found, whereas no affected patients carried a N34S mutation. In the nuclear family B (A16V trypsinogen mutation) both the patient and her healthy father carried identical PRSS1 and SPINK1 mutations. In family C (R112H trypsinogen mutation) the N34S mutation was randomly distributed between patients and relatives irrespective of the phenotype. IDDM indicates patients with insulin dependent diabetes mellitus.
Although the question of disease penetrance largely reduces the issue to an all or nothing outcome, the effects of a modifier gene can be more subtle. We therefore studied whether HP in patients with SPINK1 mutations evolved more rapidly, and compared the ages of onset of disease in patients with and without the N34S SPINK1 mutation. Because of their doubtful biological relevance and equal distribution between carriers and patients, the four people with SPINK1 polymorphisms (3′UTR 272 C>T polymorphisms (3), novel 3′ flanking region 371 G>A polymorphism (1)) were not included in this analysis. No difference was detected in the distribution of ages at onset of disease in either group and the cumulative incidences and their regression formed parallel curves (fig 2). As the proportion of cases of pancreatitis classified as mild or severe disease was not different in either the N34S patients (mild 20%) and SPINK1 wild type patients (mild 22%), we conclude that the presence or absence of SPINK1 mutations has neither an effect on the phenotypic expression nor on the disease severity of HP. To find out whether the severity of disease is determined by the age of the patient when pancreatitis begins, we compared the cumulative incidence of patients with clinically mild and severe disease (fig 3). Again, the severity of pancreatitis did not depend on the age of onset. This means that patients who develop symptoms of HP very early in life can still experience a mild clinical course whereas those who develop pancreatitis later in adulthood are not protected against developing complications of pancreatitis.

Neither (A) the cumulative incidence of pancreatitis, nor (B) the comparison of ages at onset of disease of patients with PRSS1 associated HP who carried SPINK1 wild type or the N34S mutation indicated an effect of SPINK1 on the clinical disease phenotype.

Phenotypically mild and severe cases of HP were randomly distributed between different age groups at onset of disease. Neither (A) the cumulative incidence, nor (B) the comparison of the ages at onset resulted in significant differences.
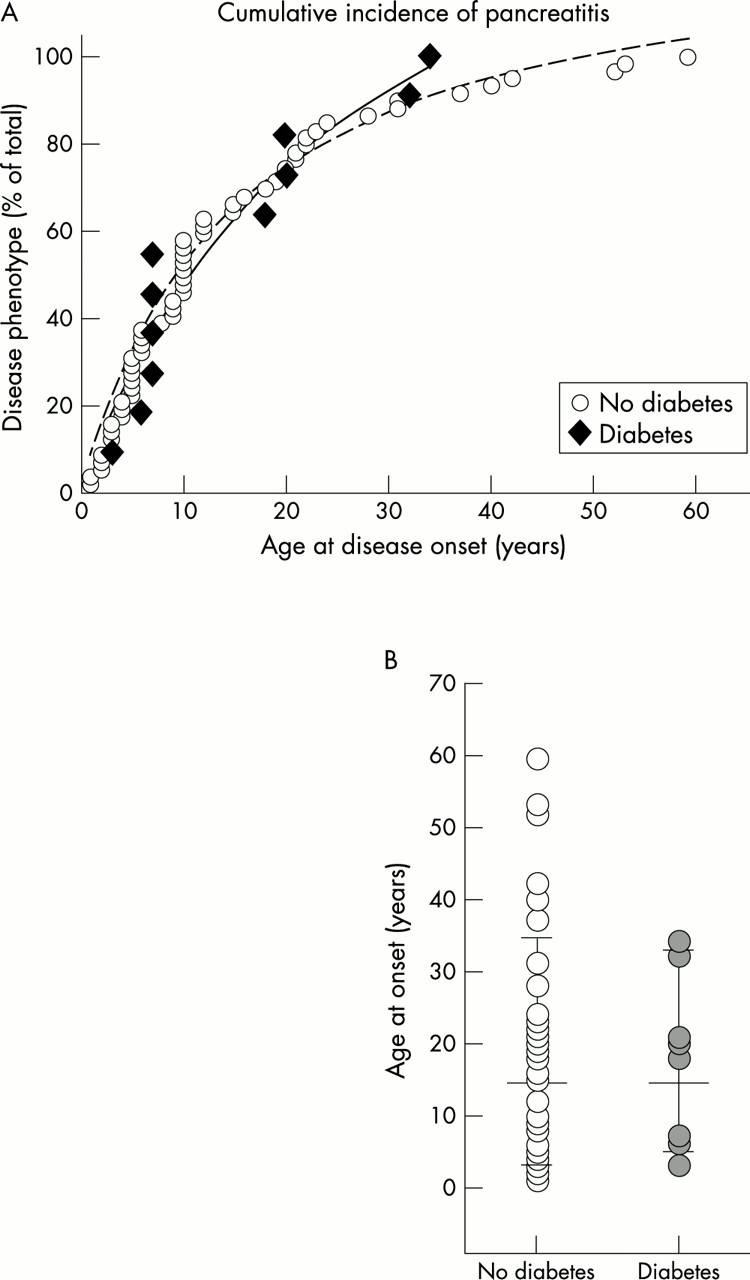
In the group of patients with pancreatitis with insulin dependent diabetes (11 in all), one carried the N34S SPINK1 mutation, one a 3’UTR 272 C>T polymorphism, and the remaining nine were SPINK1 wild type carriers. The median age at which patients developed diabetes was 37 years (mean 38 years; 95% confidence interval (95% CI) 10.3). Interestingly, two patients (7 and 34 years old) presented with diabetes as the initial symptom of HP. In the remaining patients the median duration between the onset of pancreatitis and the onset of diabetes was 20 years (mean 28; 95% CI 12.7). As an onset of disease early in childhood could increase the overall risk of patients developing diabetes, we studied the cumulative incidence of pancreatitis in patients with and without diabetes, and surprisingly found no correlation (fig 4). This result indicates that in patients with HP the development of secondary diabetes is independent of the age at which pancreatitis begins and is determined by other, as yet unknown factors. A possible exception to that rule could be found in the rare patients who phenotypically develop HP after the age of 35 years. Although none of the subjects in our cohort with an onset of disease after the age of 35 was diabetic, we cannot at present rule out that they may develop diabetes at an advanced age. This possibility also remains because the median follow up period from the onset of pancreatitis to inclusion in the study was 7 years (mean 11; 95% CI 2.9) for patients with non-diabetic pancreatitis, and thus was significantly shorter than the interval between the onset of pancreatitis and onset of diabetes in diabetic patients (p<0.005).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neither (A) the cumulative incidence of pancreatitis, nor (B) the comparison of ages at onset of disease suggested that patients with an earlier onset of disease are at higher risk of developing secondary diabetes than patients in whom pancreatitis began later in life. Whether or not patients in whom pancreatitis was diagnosed after the age of 35 will still develop diabetes at an advanced age needs to be confirmed by long term follow up.
In connection with the severity data our findings imply that patients or their doctors cannot easily predict whether a patient with HP will have a mild or severe clinical course of the disease and whether or not the patient will develop secondary diabetes. Predictions of this kind should not be based on the age at which the first symptoms of pancreatitis occur or at which the disease is diagnosed.
CONCLUSIONS
In summary, our data do not rule out that SPINK1 mutations represent a modifier for the onset of pancreatitis in general, and they do not contribute to the ongoing discussion of whether SPINK1 mutations cause idiopathic pancreatitis directly or only modify other, as yet undetermined, risk factors.14,15 In HP associated with cationic trypsinogen gene mutations, the presence or absence of SPINK1 alterations neither affects the penetrance, nor the disease severity, or the onset of secondary diabetes. For the clinical care of patients with HP this study implies that whether or not a patient develops pancreatitis as a child or as an adult, and whether or not the patient has coinherited SPINK1 mutations, will not affect his or her individual clinical course or the risk of developing insulin dependent diabetes mellitus.
Acknowledgments
The first two authors contributed equally to this work. This study was supported by the Deutsche Forschungsgemeinschaft (Le 625/4-3, Le 625/5-2, Wi 2036/1-1), the Interdisciplinary Center for Clinical Research Münster (IZKF D21), the Sonnenfeld-Stiftung, and the European Registry for Hereditary Pancreatitis and Familial Pancreatic Cancer (EUROPAC) in Liverpool and EUROPAC Germany in Münster. S Greiner and H Baumhöver provided expert technical assistance, W Greenhalf and N Howes helpful discussions, and D Stocken expert statistical advice.