Article Text

Abstract
Objective Deregulation of the Wnt signalling pathway by mutations in the Apc or β-catenin genes underlies colorectal carcinogenesis. As a result, β-catenin stabilises, translocates to the nucleus, and activates gene transcription. Intestinal tumours show a heterogeneous pattern of nuclear β-catenin, with the highest levels observed at the invasion front. Activation of receptor tyrosine kinases in these tumour areas by growth factors expressed by surrounding stromal cells phosphorylate β-catenin at tyrosine residues, which is thought to increase β-catenin nuclear translocation and tumour invasiveness. This study investigates the relevance of β-catenin tyrosine phosphorylation for Wnt signalling and intestinal tumorigenesis in vivo.
Design A conditional knock-in mouse model was generated into which the phospho-mimicking Y654E modification in the endogenous β-catenin gene was introduced.
Results This study provided in vivo evidence that β-cateninE654 is characterised by reduced affinity for cadherins, increased signalling and strongly increased phosphorylation at serine 675 by protein kinase A (PKA). In addition, homozygosity for the β-cateninE654 targeted allele caused embryonic lethality, whereas heterozygosity predisposed to intestinal tumour development, and strongly enhanced Apc-driven intestinal tumour initiation associated with increased nuclear accumulation of βcatenin. Surprisingly, the expression of β-cateninE654 did not affect histological grade or induce tumour invasiveness.
Conclusions A thus far unknown mechanism was uncovered in which Y654 phosphorylation of β-catenin facilitates additional phosphorylation at serine 675 by PKA. In addition, in contrast to the current belief that β-catenin Y654 phosphorylation increases tumour progression to a more invasive phenotype, these results show that it rather increases tumour initiation by enhancing Wnt signalling.
- β-Catenin
- colorectal cancer
- molecular biology
- mouse model
- tyrosine 654 phosphorylation
- Wnt signalling
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Increased Wnt/β-catenin signalling underlies the initiation and progression of colorectal cancer.
Receptor tyrosine kinases are activated in colorectal cancer by growth factors coming from the tumour stroma.
Receptor tyrosine kinases can phosphorylate tyrosine 654 of β-catenin.
What are the new findings?
We provide in-vivo evidence that tyrosine 654 phosphorylated β-catenin shows reduced interaction with cadherins, increased signalling and strongly increased phosphorylation at serine 675 by protein kinase A (PKA).
Expression of tyrosine 654 phosphorylated -catenin predisposes to intestinal tumour development in mice.
Tyrosine 654 phosphorylation of β-catenin strongly enhances Apc-driven intestinal tumour initiation, but not progression to a more aggressive tumour phenotype.
How might it impact on clinical practice in the foreseeable future?
Our results suggest that therapy targeted at the tumour stroma, tyrosine kinases and PKA, may serve an efficient treatment strategy for patients with intestinal or other cancers involving active Wnt signalling.
Most cases of sporadic colorectal cancer are initiated by activation of the Wnt/β–catenin signalling pathway through mutations in either adenomatous polyposis coli (APC) or β-catenin itself. β-Catenin is involved in cell adhesion through forming cadherin–catenin complexes as well as in gene transcription through binding to TCF/LEF as a co-activator of transcription. N-terminal serine/threonine phosphorylation of β-catenin by GSK3 and CK1 targets it for degradation, whereas inactivation of APC leads to cytoplasmic accumulation of β-catenin, which eventually translocates to the nucleus to activate transcription.1 This model implies that nuclear accumulation of β-catenin should earmark all APC-deficient cells; however, a heterogeneous pattern of nuclear β-catenin is observed in APC-driven intestinal tumours.2 3 This indicates that additional mechanisms are involved in β-catenin nuclear translocation and in the regulation of Wnt signalling activation by modulating β-catenin.4 Nuclear β-catenin staining strongly correlates with tumour size and the grade of dysplasia, with the highest levels of nuclear β-catenin at the invasion fronts of adenocarcinomas.2 3 For these reasons, obtaining a better understanding of the processes that control nuclear accumulation of β-catenin during tumour progression is of great clinical relevance.
One of the mechanisms affecting Wnt signalling is β-catenin tyrosine (Y) phosphorylation by active receptor tyrosine kinases (RTK).5 6 Tumour cells as well as stromal cells in the tumour microenvironment secrete growth factors that activate their cellular receptors expressed at the plasma membrane of tumour cells. In a large number of cancers, including colorectal cancer, overexpression of RTK or expression of structurally altered RTK results in increased growth factor signalling.7 Upon activation of RTK, various downstream adaptor and signalling molecules are recruited and phosphorylated, thus triggering the activation of different intracellular pathways, among which are the RAS/MAPK and the PI3K/AKT pathways.7 In addition, RTK activation leads to tyrosine phosphorylation of β-catenin affecting its functions in cell adhesion and signalling. In particular, phosphorylation of β-catenin residue Y654 results in its release from cadherins and an increase in TCF-mediated transcriptional activity.8–11 Altogether, these results suggest that β-catenin Y654 phosphorylation balances its role between cell adhesion and Wnt signalling.5 6 8 As such, enhanced Y654 phosphorylation of β-catenin is believed to increase cell migration and induce an invasive behaviour of tumour cells, although functional evidence for the latter is still lacking.
To determine the relevance of β-catenin Y654 phosphorylation for Wnt signalling and intestinal tumorigenesis in vivo, we have generated a conditional knock-in mouse model in which we introduced the phospho-mimicking Y654E modification in the endogenous β-catenin gene. The resulting β-cateninE654 protein is characterised by reduced affinity for cadherins and increased transcriptional activity. We show that while homozygosity for the β-cateninE654 targeted modification results in embryonic lethality, heterozygosity for β-cateninE654 is sufficient to initiate intestinal tumour formation and to increase Apc-driven intestinal tumour initiation. Importantly, these phenotypes are not accompanied by an increased migratory phenotype, showing that, in contrast to current belief, β-catenin Y654 phosphorylation by itself is not sufficient to impose an invasive phenotype onto cells. Moreover, we uncovered a thus far unknown mechanism in which Y654 phosphorylation of β-catenin facilitates additional phosphorylation at serine (S) 675 by protein kinase A (PKA). Taken together, we provide in-vivo evidence that β-catenin Y654 phosphorylation enhances Wnt signalling and thereby increases intestinal tumour formation.
Materials and methods
Mouse strains
The mouse strains (C57BL/6J genetic background) used in this study were: Apc+/15lox, Apc+/1572T, Apc+/1638N, Fabpl-Cre, CAG-Cre and CAG-Flp.12–17 All mice were generated, bred and maintained under specific pathogen-free conditions at the animal facility of the Erasmus Medical Centre (Rotterdam, The Netherlands). All animal experiments were approved by the Institute's Animal Ethics Committee and were carried out in accordance with Dutch and international legislation.
Generation of conditional β-cateninY654E knock-in mice
A β-catenin targeting vector was constructed as described in the supplementary material available online only. One targeted ES clone was used to obtain chimeras by injecting C57BL/6J blastocysts and breeding with C57BL/6J mice to produce outbred heterozygous β-cateninY654/Y654E-fl-neo mice (mixed C57BL/6J;129Ola background). These mice were mated with either CAG-Flp or CAG-Cre mice to obtain β-cateninY654/E654-fl and β-cateninY654/E654 mice, respectively. Mice and embryos were genotyped as described in the supplementary material, available online only.
Skeletal preparations and staining
β-cateninY654/E654 mice were backcrossed to a C57Bl6/J genetic background (seven generations) and interbred. Embryos of E15.5 were harvested, skinned and eviscerated, fixed overnight in 96% ethanol containing 1% glacial acetic acid. Cartilage was stained overnight in 0.5 mg/ml Alcian Blue. Soft tissue was dissolved in 1.5% potassium hydroxide for 3 h. Bone was then stained overnight in 0.15 mg/ml Alizarin Red. Embryos were destained in 0.5% potassium hydroxide/20% glycerol for 3 days.
Intestinal tumour analysis
The entire intestines were isolated, opened longitudinally, washed in phosphate-buffered saline and fixed overnight at 4°C in 10% phosphate-buffered formalin. The size and location of tumours was determined using a dissection microscope.
Tissue processing and immunohistochemical analysis
Fixed intestinal tumours and embryos (4% paraformaldehyde) were routinely embedded in paraffin. Sections were stained by haematoxylin eosin for routine histology. All tumours were histologically classified according to previously reported criteria.18 Primary and secondary antibodies employed for immunohistochemical analysis are depicted in the supplementary material, available online only. Proliferation rates were quantified by determining the average percentage of phosphoHistone-H3 positive cells in five microscopic fields per tumour. Percentages of cells positive for nuclear β-catenin were determined as an average of five microscopic fields per tumour, and were scaled in three groups, less than 25%, 25–50% or over 50% of tumour cells with nuclear β-catenin.
Allelic imbalance analysis
Tumour (T) and normal (N) tissue was microdissected from three paraffin sections and DNA was isolated as described previously.15 PCR were performed using the geno654-F and geno654-R primers (supplementary material, available online only) to show the presence of the Y654 and E654 alleles.
Cell culture, transient transfection and treatments
β-cateninY654/E654 mice were backcrossed to a C57BL/6J genetic background (seven generations) and interbred. Mouse embryonic fibroblasts (MEF) were isolated from embryos of E13.5–15.5 and cultured as previously described.19 Cos-1 cells were cultured in Dulbecco's modified essential medium complemented with 1% P/S and 10% fetal bovine serum at 37°C, 5% carbon dioxide. MEF and Cos-1 cells were transfected using FuGENE HD (Roche) or Polyethylenimine Max (PEI; Polysciences, Inc, Eppelheim, Germany), respectively. Plasmids used in Cos-1 cells were pEGFP-C1 GFP-β-catenin expression vectors, in which the Y654E and Y654F mutations were introduced by site-directed mutagenesis. Cells were treated with 25 μM H89 (Enzo Life Sciences, Lausen, Switzerland) for 24 h or 50 μM forskolin (Merck, Whitehouse Station, NJ, USA) for 1 h.
β-Catenin reporter assay
β-Catenin reporter assays were performed in MEF using the Wnt-responsive element-luciferase reporter kindly provided by Dr Georges Rawadi (Galapagos, Paris, France) for which we generated a mutant-responsive element-luciferase variant. Tk-Renilla was used as a control for transfection efficiency. After transfection, cells were stimulated for 24 h with 25% of l-control or l-Wnt3A-conditioned medium.20 Luciferase activities were measured using the dual-luciferase reporter assay system (Promega, Madison, WI, USA) and a LUMIstar luminometer according to the manufacturer's protocol.
Immunoprecipitation and fluorescent immunoblotting
Cell lysates for co-immunoprecipitation (coIP) were generated using E1A buffer (50 mM Tris pH 7.5, 250 mM NaCl, 5 mM ethylene diammine tetraacetic acid, 0.1% Nonidet-P40, complete protease inhibitor cocktail (Roche, Basel, Switserland), 1 mM sodium vanadate (Na3VO4), 1 mM phenyl-methylsulphonyl fluoride). Protein concentrations were determined using Quickstart Bradford (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol; 100 μg of protein was incubated with 0.25 μg β-catenin antibody (BD Transduction Laboratories, Franklin Lakes, NJ, USA) overnight at 4°C and precipitated using protein G Sepharose beads (GE Healthcare, Chalfont St Giles, UK). Precipitated proteins were resuspended in 100 μl Laemmli sample buffer. Fluorescent Odyssey immunoblotting (LI-COR Biosciences, Lincoln, NE, USA) was performed for these immunoprecipitated proteins and total lysates as described in the supplemental materials.
Results
Homozygosity for β-cateninE654 results in embryonic lethality
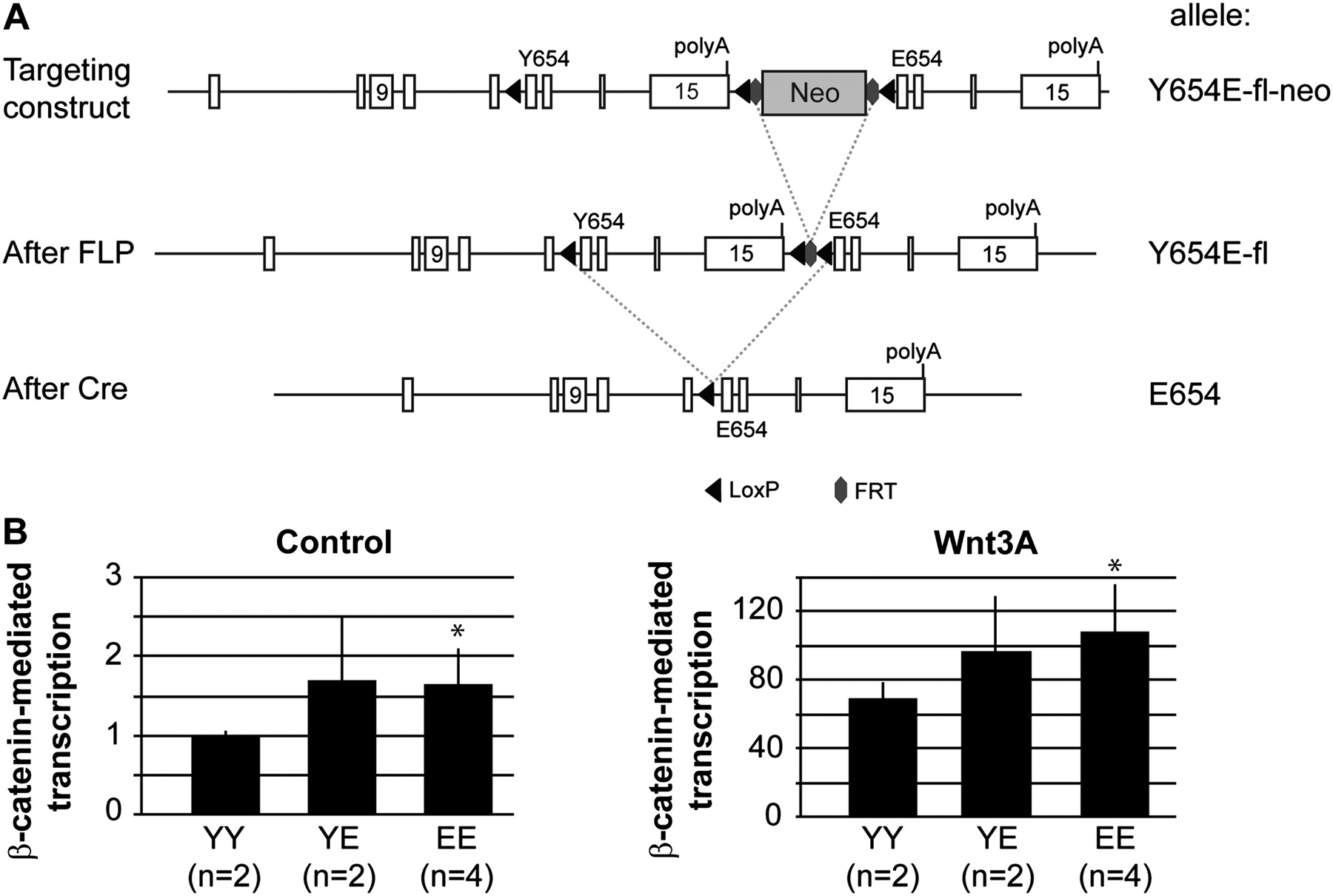
We generated a conditional β-catenin knock-in mouse model in which, following Cre activity, exons 12–15 enclosing Y654 will be replaced by the same exons containing a glutamic acid (E) at residue 654, that is β-cateninY654/Y654E-fl mice (figure 1A). Heterozygous β-cateninY654/Y654E-fl mice were bred with CAG-Cre mice to obtain heterozygous β-cateninY654/E654 mice, expressing both the β-cateninY654 and β-cateninE654 alleles at equal transcriptional levels (see supplementary figure S1A, available online only). These animals were interbred to obtain several independent MEF lines expressing either wild-type β-catenin (YY), β-cateninE654 (EE), or expressing both forms of β-catenin (YE) at endogenous levels. Using both control and Wnt3A ligand-stimulated conditions, we measured a significant increase (p<0.005) in β-catenin-mediated transcription in MEF expressing β-cateninE654 (figure 1B), indicating that the β-catenin Y654E modification increases Wnt signalling in vivo.

Generation of β-cateninY654/E654 mice. (A) A Y654E-fl-neo allele was generated by homologous recombination in ES cells, using the targeting vector described in the supplementary material, available online only. Breeding with CAG-Flp mice removes the Neo cassette resulting in the Y654E-fl allele. By breeding with CAG-Cre or Fapbl-Cre mice, the original exons 12–15 were removed, ending up with the E654 allele. (B) β-Catenin reporter assays were performed to measure β-catenin-mediated transcription in mouse embryonic fibroblasts (MEF) expressing β-cateninY654/Y654 (YY), β-cateninY654/E654 (YE) or β-cateninE654/E654 (EE). Following transfection, β-catenin signalling was induced by stimulating MEF with Wnt3A ligand. Wnt-responsive element/mutant-responsive element ratios were measured in three individual experiments in duplo, and averaged. *p<0.005, Student's t-test. Numbers between brackets refer to the number of independent cell lines used for each genotype. Cre, Cre recombinase; FLP, flippase recombinase; FRT, FLP recombinase target; LoxP, Cre recombinase target.
Interbreeding of β-cateninY654/E654 animals showed that homozygosity leads to embryonic lethality around birth (see supplementary table S1, available online only). Macroscopic analysis of whole embryos at E10.5 revealed that homozygous β-cateninE654/E654 embryos had smaller head sizes and unfused facial processes, both typical features of anterior truncation (figure 2A). We did not observe any additional deformities in the remainder of the embryos, nor could we observe malformations in heterozygous β-cateninY654/E654 embryos (figure 2A). Sagittal sections of E12.5 homozygous β-cateninE654/E654 embryos showed that the most rostral regions of the head were severely underdeveloped (figure 2B). The primordia of the nasal capsule and upper jaw were highly abnormal or absent, whereas the development of mandible and tongue was only mildly affected. Skeletal stainings of E15.5 embryos showed that the dermal skull roof as well as the upper jaw were virtually absent, while the lower jaw (Meckel's cartilage and dentaries) appeared to be near normal (figure 2C). These data indicate that the increase in Wnt signalling resulting from the expression of β-cateninE654 causes an embryonic lethal phenotype similar to phenotypes observed in other Apc and Wnt-related mouse models.21–23
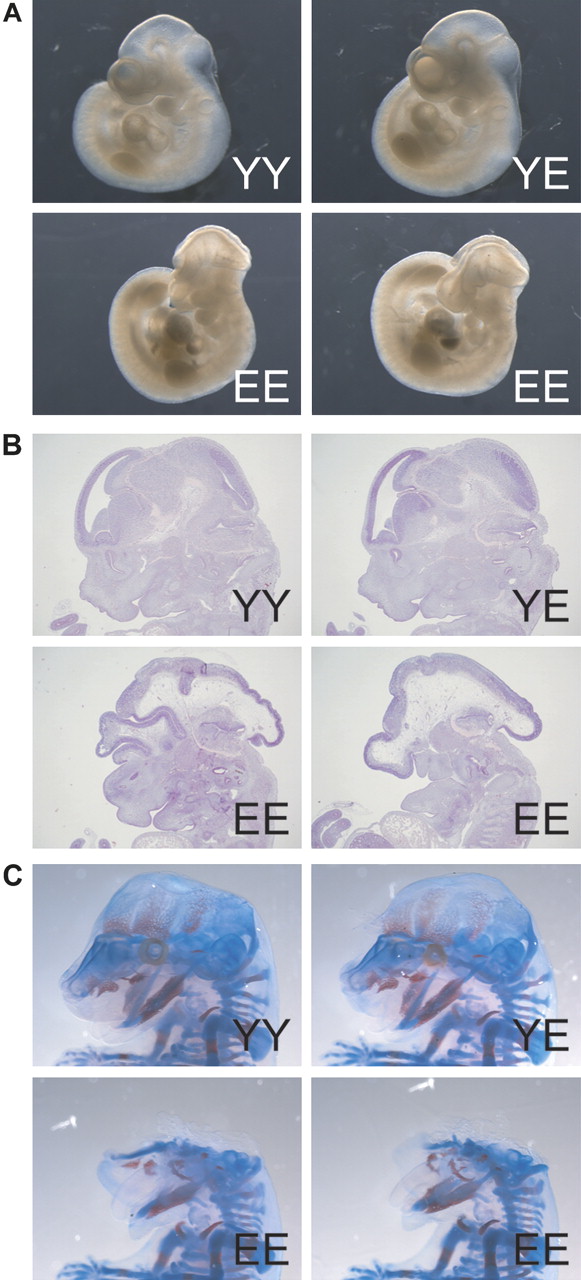
Anterior truncation in homozygous β-cateninE654/E654 embryos. (A) Whole embryo pictures at E10.5 showing a severe anterior truncation phenotype of homozygous β-cateninE654/E654 (EE) embryos compared to wild-type (YY) and heterozygous β-cateninY654/E654 (YE) embryos. (B) Haematoxylin eosin stainings of sagittal sections of the head region of E12.5 embryos. (C) Skeletal stainings of E15.5 embryos.
Despite reduced affinity for cadherins, β-cateninE654 is still localised at the plasma membrane
Previously reported transient overexpression experiments suggested that the interaction between β-catenin and cadherins was reduced by the Y654E modification,9–11 resulting in a switch of β-catenin from the membranous adhesion pool to the nuclear signalling pool. To investigate whether this was also the case for β-cateninE654 at endogenous levels, we performed coIP experiments using our MEF lines. Protein expression levels of the β-cateninE654 variant were similar to β-cateninY654, and we observed no clear differences in the expression of cadherins between the different MEF lines (figure 3A). However, the interaction of β-cateninE654 with cadherins was clearly impaired compared with β-cateninY654, suggesting that the involvement of β-cateninE654 in adhesion junctions is reduced (figure 3A,B).

β-cateninE654 in cadherin-catenin complexes. (A) Total endogenous β-catenin was immunoprecipitated from YY, YE and EE mouse embryonic fibroblasts (MEF). Co-precipitated cadherins were detected by immunoblotting with two individual antibodies (Ncad and PANcad). Expression levels of β-catenin as well as of cadherins were shown in total lysates (input). (B) Quantification of interaction between β-catenin and cadherins. The interactions were calculated by correcting the amount of co-precipitated cadherins for the amount of precipitated β-catenin. The interaction levels were quantified in three individual experiments, and averaged. Numbers between brackets refer to the number of independent cell lines used for each genotype. (C) Representative E-cadherin immunostaining of bronchial alveoli of a wild-type and homozygous β-cateninE654/E654 embryo (400×). (D) Representative β-catenin immunostaining of epidermis of a wild-type and homozygous β-cateninE654/E654 embryo (400×).
Although the reduced interaction between β-cateninE654 and cadherins suggested that cell adhesion would be affected, the formation of epithelial structures in β-cateninE654/E654 embryos was not impaired, and a normal expression pattern for E-cadherin could be detected in all epithelia (figure 3C). Notably, β-catenin was also detected at the plasma membrane at equal or, at most, slightly reduced levels in epithelia of β-cateninE654/E654 embryos, and could not be observed in the cytosol or nuclei of epithelial cells (figure 3D), suggesting that the Y654E modification is not sufficient to shift β-catenin from the plasma membrane to the nucleus, at least not at levels detectable with immunohistochemistry. Overall, these data indicate that increased Wnt signalling by the expression of β-cateninE654 causes anterior malformations during embryonic development, without affecting the formation of epithelial structures.
Heterozygous β-cateninY654/E654 mice develop intestinal adenomas
As β-cateninE654 increases Wnt signalling, we questioned whether the expression of β-cateninE654 would predispose to tumour development in vivo. Therefore, we examined heterozygous β-cateninY654/E654 mice and wild-type littermates at the age of 16-18 months for abnormalities with a specific focus on lesions typically developing in Apc mutant models, that is intestinal and mammary tumours, cutaneous cysts and desmoids.13–15 19 24 25 Fourteen tumours were identified in the duodenum or jejunum of 11 out of 29 (38%) heterozygous animals compared with none out of nine wild-type littermates (p=0.038) (figure 4A). Fifty per cent of these intestinal tumours were histologically identified as gastrointestinal intraepithelial neoplasia/adenoma, 21% as adenoma and 29% as dysplastic adenoma/carcinoma in situ according to previously described criteria.18 We did not observe any predisposition for other lesions such as cysts, desmoids or mammary tumours.
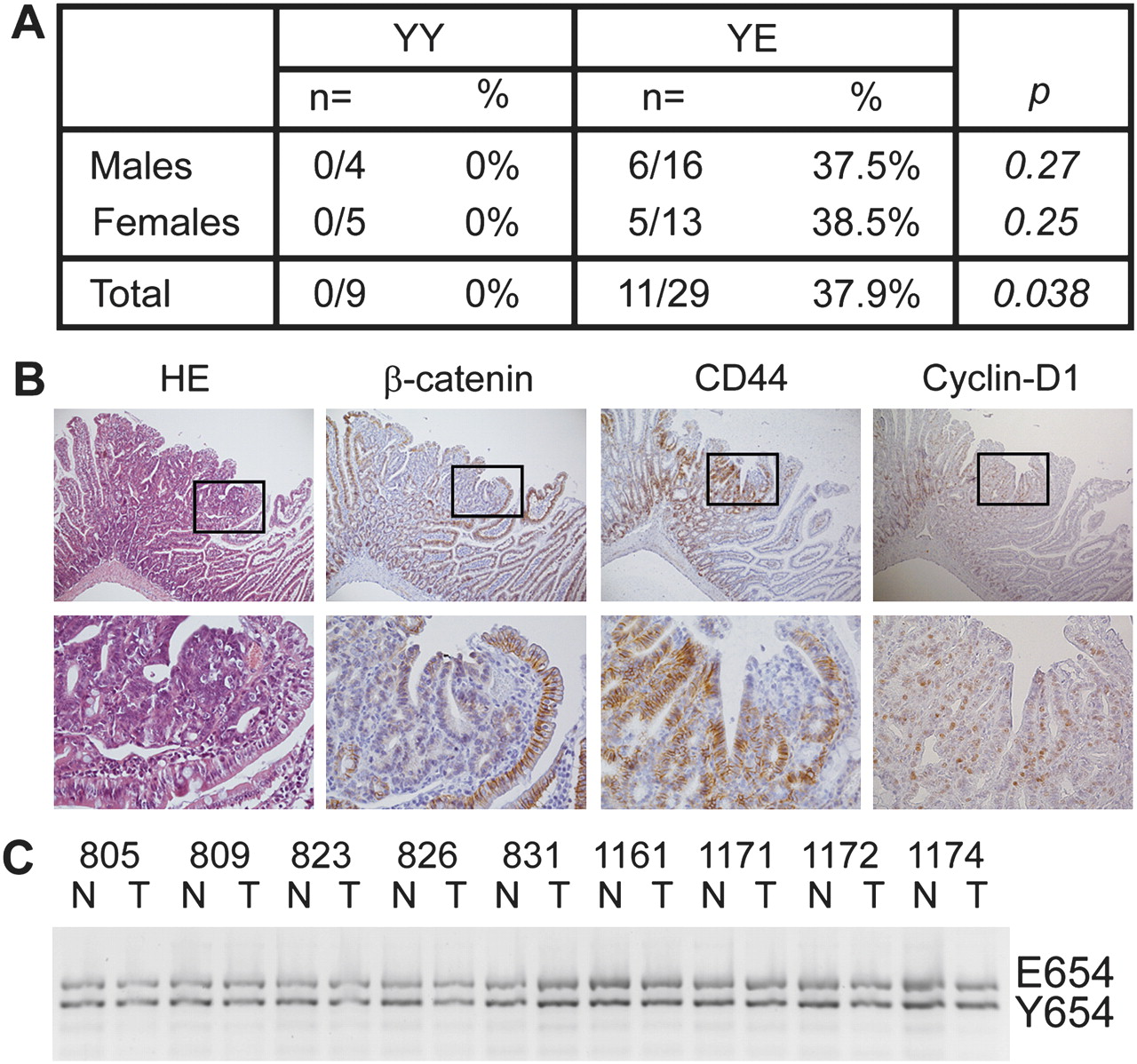
Heterozygous β-cateninY654/E654 mice develop intestinal tumours. (A) Numbers and percentages of wild-type (YY) and heterozygous β-cateninY654/E654 (YE) mice that have developed at least one intestinal tumour at the age of 16–18 months. p Values, Fisher's exact test. (B) Haematoxylin eosin staining, and β-catenin, CD44 and cyclin-D1 immunostainings of a representative intestinal tumour of a β-cateninY654/E654 mouse. Membranous β-catenin levels were strongly reduced in tumour cells compared with normal intestinal epithelial cells, while no increase in cytoplasmic/nuclear β-catenin could be observed. The expression of both CD44 and cyclin-D1 was increased in tumour cells. Lower panels are higher magnifications (400×) of the boxes indicated in the upper panels (100×). (C) Allelic imbalance analysis revealed no loss of either the Y654 or E654 alleles in intestinal tumours of β-cateninY654/E654 mice investigated. N, normal tissue, T, tumour tissue.
Immunohistochemical analyses revealed that β-catenin expression was strongly reduced at the plasma membrane of tumour cells compared with normal intestinal epithelial cells (figure 4B); however, nuclear β-catenin could not be observed (figure 4B). Allelic imbalance analysis revealed that both the wild-type (Y654) and modified (E654) alleles were retained in all nine intestinal tumours tested (figure 4C). Although nuclear β-catenin could not be observed in the tumour cells, the expression of CD44 and cyclin-D1, both canonical β-catenin target genes,26 was increased in these tumours (figure 4B). These results suggest that the expression of β-cateninE654 predisposes to intestinal tumour formation associated with increased Wnt signalling.
Embryonic lethality of compound heterozygous β-catenin;Apc mice
To investigate the effect of β-cateninE654 on Apc-driven tumour formation, we bred our β-catenin model with two mutant Apc knock-in mouse models, that is Apc+/1572T and Apc+/1638N.13–15 However, compound heterozygous β-catenin;Apc animals could not be obtained from these breedings, indicating that heterozygosity for the Apc1572T or Apc1638N mutations acts synergistically with the heterozygous Y654E modification in β-catenin to induce an embryonic lethal phenotype (see supplementary table S2, available online only). Compound heterozygous β-catenin;Apc embryos showed an anterior truncation phenotype similar to the one observed in β-cateninE654/E654 embryos (see supplementary figure S2, available online only). The telencephalic region of β-cateninY654/E654;Apc+/1638N embryos was more severely reduced compared with β-cateninY654/E654;Apc+/1572T embryos. Accordingly, β-cateninY654/E654 embryos in combination with the Apc1638N mutation were less commonly detected compared with the ones combined with the Apc1572T mutation, 11.1% and 29.7%, respectively (see supplementary table S2, available online only). These results suggest that the proper development of mice expressing the β-cateninE654 allele in combination with the Apc1638N allele is more severely affected compared with the Apc1572T allele, which is probably explained by our previous observations that Apc1572T retains more residual β-catenin downregulating activity than Apc1638N.13 19
In conclusion, the lethality observed in these compound heterozygous embryos indicates that retaining only half the level of functional Apc is insufficient to counterbalance the increased propensity of β-cateninE654 to signal to the nucleus, and confirms our observation that the β-cateninE654 allele contributes to increased Wnt signalling.
β-CateninE654 increases Apc-driven intestinal tumour formation
The embryonic lethality of compound Apc;β-catenin mice, described above, precludes the analysis of tumorigenesis in adult phenotypes. Therefore, we generated intestinal-specific Apc;β-catenin double mutant mice to determine whether β-cateninE654 contributes to Apc-driven intestinal tumorigenesis. To this aim, we used the Fabpl-Cre mouse model, which expresses Cre recombinase specifically in the ileum, cecum and large intestine.16 We bred Fabpl-Cre mice with conditional β-cateninY654/Y654E-fl-neo mice and with conditional Apc+/15lox mice12 in order to generate compound heterozygous Apc+/Δ15;β-cateninY654/E654 mice expressing both Apc and β-catenin variants specifically in the intestines. At 8 months of age, Apc+/Δ15;β-cateninY654/E654 mice (from here on Apc;YE mice) showed a significant increase in tumour number compared with Apc+/Δ15;β-cateninY654/Y654 mice (from here on Apc mice) (p=0.013). Notably, tumour numbers were significantly increased in males (p=0.02), but not in females (p=0.48) (figure 5A). Age-matched Apc+/+;β-cateninY654/E654 mice (from here on YE mice) and other control littermates had not developed intestinal tumours. Tumours of Apc mice were localised predominantly in the ileum, but also in the cecum and large intestine (figure 5B). Upon additional expression of the β-cateninE654 variant, the distribution of tumours was shifted more towards the large intestines, both in males and females (figure 5B). These data show that heterozygosity for the β-cateninE654 targeted allele increases Apc-driven intestinal tumour formation and shifts tumour location.

Intestinal tumour incidence of intestinal-specific Apc and Apc;YE mice. (A) Total amount of tumours detected in intestinal-specific Apc and Apc;YE mice at the age of 8 months. Black lines indicate the median amount of tumours per genotype group. *p=0.02, Mann–Whitney test. (B) Percentage of intestinal tumours detected in ileum, cecum or large intestine of Apc and Apc;YE mice. (C) Percentages of cells of tumours from male Apc and Apc;YE mice showing nuclear β-catenin. CIS, carcinoma in situ. (D) Histological classification of intestinal tumours of Apc and Apc;YE mice according to Boivin et al.18 (E) Average diameter in mm of all intestinal tumours of Apc and Apc;YE mice. *p=0.022, **p=0.0001, Student's t-test.
Because Y654 phosphorylation of β-catenin leads to reduced interaction with cadherins and increased Wnt signalling, current belief is that this would result in increased tumour invasiveness.6 To investigate whether β-cateninE654 contributes to Apc-induced tumour progression in vivo, we determined the histological grade, size and proliferation rate of tumours from male Apc and Apc;YE mice. The tumours were histologically classified as hyperplasia/adenoma, dysplastic adenoma/carcinoma in situ or adenocarcinoma, but no differences in malignancy could be observed between both groups (figure 5C). Tumours of Apc;YE mice were on average smaller compared with tumours from Apc mice, both for males (p=0.0001) and females (p=0.022) (figure 5D and supplementary figure S3A, available online only). PhosphoHistone-H3 stainings of size-matched tumours from male Apc;YE mice showed a slightly although not significant (p=0.09) increase in the proliferation rate compared with tumours from Apc mice (see supplementary figure S3B, available online only). These results indicate that β-cateninE654 increased Apc-driven intestinal tumour initiation, but not tumour progression.
Because tumours of Apc+/Δ15 mice show a clear nuclear accumulation of β-catenin,12 we hypothesised that the introduction of the Y654E modification in β-catenin would further increase the percentage of cells showing nuclear β-catenin. Immunohistochemical analysis revealed that β-catenin was more predominantly localised in the nuclei of Apc;YE tumours compared with Apc tumours (figure 5E). Together, this indicates that β-cateninE654 translocates more readily to the nuclear signalling pool of Apc-deficient cells in vivo, thereby contributing to Apc-driven tumour initiation without clearly affecting tumour invasion and metastasis.
Phosphorylation at β-catenin Y654 facilitates phosphorylation at S675 by PKA
Xing et al27 described that the armadillo repeat 12 in which Y654 is located forms several H-bonds with the C-tail of β-catenin. Residues up and downstream of Y654, that is Leu644 and Phe660, directly interact with residues in the C-tail of β-catenin, among which are leucine residues at codons 674 and 678.27 In between these two leucine residues, another phosphorylation site of β-catenin is located, that is S675.28 Piedra et al10 have suggested that the S675-containing C-tail of β-catenin folds open upon phosphorylation of Y654, due to the negative charge introduced at armadillo repeat 12. This led us to hypothesise that, upon phosphorylation of Y654, the S675 becomes available for additional phosphorylation. Therefore, we determined the levels of S675-phosphorylated β-catenin in total lysates of the MEF lines, using an antibody specifically recognising phosphoS675 β-catenin.29 Notably, phosphoS675 β-catenin levels were clearly increased in EE-MEF (figure 6A). The same results were detected in total lysates of β-cateninE654/E654 embryos (figure 6B), and in intestinal tumours from Apc;YE mice (figure 6C).

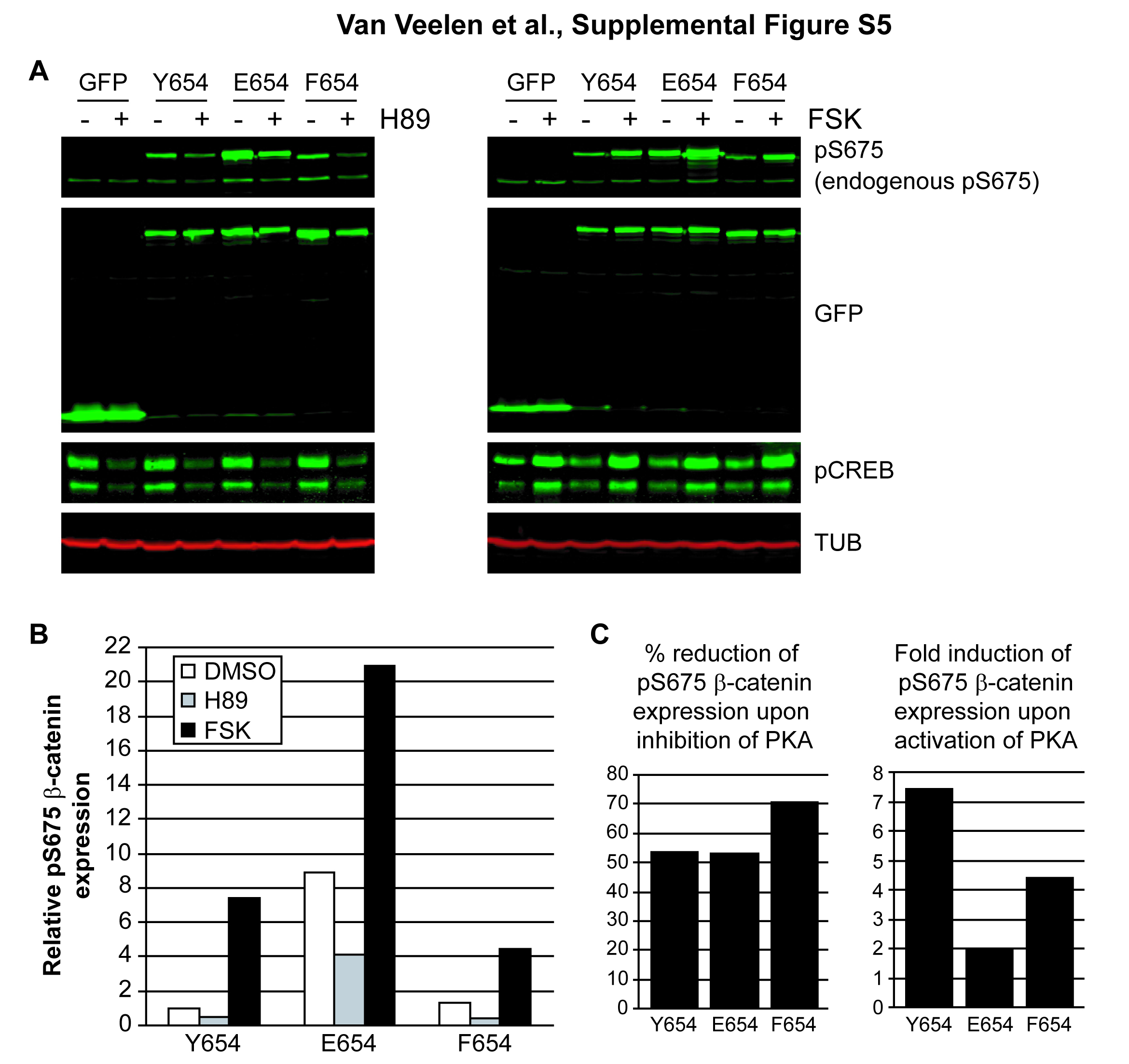
Phosphorylation at S675 in β-cateninE654 by protein kinase A (PKA). (A–C) Immunoblotting showing total β-catenin (βcat) and phosphoS675 β-catenin (pS675) expression levels in total lysates of wild-type (YY), β-cateninY654/E654 (YE) and β-cateninE654/E654 (EE) mouse embryonic fibroblasts (MEF) (A), embryos (B) and intestinal tumours from Apc and Apc;YE mice (C). Tubulin (TUB) expression levels were used as a loading control. The relative expression levels were determined in three individual experiments and averaged. Numbers between brackets refer to the number of independent cell lines used for each genotype. (D, E) MEF were treated with H89 (D) or forskolin (FSK) (E). Effects on PKA activity were evaluated by determining the expression levels of phosphoCREB (pCREB, upper band; lower band is phosphoATF-1). Following treatment, phosphoS675 β-catenin expression levels were determined and quantified. DMSO, dimethyl sulphoxide.
S675 is suggested to be a phosphorylation site for PKA.28–30 To confirm that PKA is the main kinase phosphorylating S675 in β-cateninE654, we incubated the MEF lines with a PKA inhibitor (H89) or PKA activator (forskolin). The efficiency of these agents on PKA activity was confirmed by decreased or increased phosphorylation of CREB, a specific PKA substrate (figure 6D,E). Upon inhibition of PKA, reduced levels of phosphoS675 β-catenin could be observed in all MEF lines, indicating that PKA is, at least partly, responsible for S675 phosphorylation of β-catenin (figure 6D). Upon activation of PKA, phosphoS675 β-catenin levels were increased in all MEF lines, although proportionally to a lower extent in the EE-MEF when compared with their wild-type controls (figure 6E). We could not observe a difference in PKA activity between the different MEF lines, indicating that PKA activity is not altered by the expression of β-cateninE654 (see supplementary figure S4, available online only). Therefore, we hypothesised that the Y654E modification in β-catenin facilitates phosphorylation of S675 by PKA.
Next, we transiently transfected Cos-1 cells with vectors expressing GFP-tagged wild-type β-catenin (Y654) or variants thereof (E654 and F654). The latter variant mimics dephosphorylated Y654, and may therefore retain a closed conformation of the C-tail.10 Similar to the MEF, phosphoS675 β-catenin levels were increased for E654 (see supplementary figure S5A and B, available online only). Notably, the phosphoS675 β-catenin level of F654 was comparable with Y654 (see supplementary figure S5A and B, available online only). PhosphoS675 β-catenin levels were again inhibited by H89, confirming that PKA is involved in β-catenin S675 phosphorylation (see supplementary figure S5A and B, available online only). Notably, S675 phosphorylation of β-cateninF654 could not be increased to the same extent as wild-type β-catenin by forskolin, indicating that S675 in β-cateninF654 is less available for phosphorylation by PKA (see supplementary figure S5C, available online only). From these data we concluded that Y654 phosphorylation of β-catenin is not required for but facilitates additional phosphorylation of S675 by PKA.
Discussion
Increased growth factor signalling activity has been observed in a large number of cancers, generally associated with poor prognosis. In many cases this is the result of the overexpression of the receptor or its ligand, whereas in other cases structural alterations of RTK are present resulting in constitutive kinase activity.31–34 This increased RTK activity triggers the activation of various intracellular pathways. Using in-vitro cell culture systems, various reports have shown that RTK activation also leads to tyrosine phosphorylation of β-catenin with subsequent loss of cell adhesion and increased cell motility characteristic of malignant tumour cells.5 8 Here, we describe the generation and functional characterisation of a conditional knock-in mouse model expressing a Y654 modified β-catenin at endogenous levels, allowing us to discern the relative contribution of β-catenin Y654 phosphorylation within the complexity of a developing tumour and organism.
The main consequence of introducing a phospho-mimicking modification at Y654 of β-catenin, that is β-cateninE654, is its increased Wnt signalling activity, which is supported by the nearly twofold increase in β-catenin reporter activity in MEF solely expressing β-cateninE654. Further support is provided by the embryonic phenotypes. Increased Wnt signalling caused by homozygosity for the β-cateninE654 allele resulted in an anterior truncation phenotype highly similar to that observed in several other mouse and Xenopus models in which Wnt signalling is enhanced.21–23 35 Moreover, retaining only half the level of functional Apc is insufficient to counterbalance the increased propensity of β-cateninE654 to signal to the nucleus, which can be concluded from the embryonic lethal phenotype imposed in mice compound heterozygous for the β-cateninE654 and mutant Apc alleles. In addition to these embryonic phenotypes, expression of the β-cateninE654 variant in Apc-mutant intestinal tissue resulted in enhanced intestinal tumour formation strongly associated with increased levels of nuclear accumulation of β-catenin, underscoring the potency of β-cateninE654 to translocate to the nucleus and increase Wnt signalling in vivo. Notably, heterozygosity for the targeted β-cateninE654 allele predisposed to intestinal tumours confirming the tumour-promoting potential of β-catenin modified by Y654 phosphorylation. The tumour phenotype in these mice showed a low penetrance and late onset, indicating that additional events are required to drive intestinal tumorigenesis sufficiently.
The increased signalling activity of β-cateninE654 can partly be explained by the strong enhancement of PKA-induced phosphorylation of S675, for which it has been shown to increase Wnt signalling by recruiting transcriptional co-activators.28–30 36
A second proposed mechanism for increased Wnt signalling is the reduced affinity of β-cateninE654 for cadherins leading to an increased cytoplasmic pool, thereby putting more pressure on the β-catenin destruction complex to control signalling levels properly.9–11 Our data support this concept, as we have observed a nearly 10-fold reduction in the binding affinity of β-cateninE654 for cadherins. However, despite this reduced affinity, β-cateninE654 localises at the plasma membrane in epithelia of homozygous β-cateninE654/E654 embryos, suggesting that β-cateninE654 is still incorporated in adherens junctions in vivo. In line with this, Tominaga et al37 previously reported that cells expressing exogenous β-cateninE654 at endogenous levels retained their adhesive capacity similar to cells transfected with wild-type β-catenin. Therefore, we conclude that although Y654 phosphorylated β-catenin has a reduced affinity for cadherins, it is still considerably involved in the regulation of cell adhesion.
Despite the fact that β-catenin Y654 phosphorylation results in a reduced interaction with cadherins and increased gene transcription, no obvious cytoplasmic or nuclear localised β-catenin could be observed in epithelia of homozygous β-cateninE654/E654 embryos using immunohistochemistry. In intestinal tumours of Apc;YE mice, increased nuclear localised β-catenin could, however, be observed, indicating that β-cateninE654 shifts more readily to the nuclear signalling pool in the absence of a functional degradation complex. The strong increase in signalling activity that we observed when cells are exposed to Wnt3a ligand shows that β-cateninE654 is still efficiently targeted for degradation. Together, this implies that, although Wnt signalling is increased, nuclear levels of β-catenin upon C-terminal phosphorylation are too low to be detected with immunohistochemistry, and become more apparent when the destruction complex is either mutated such as in Apc mutant colorectal tumours, or is temporarily inhibited by extracellular Wnt signals such as is the case in anterior embryonic development.
Because of its dual role in cell adhesion and Wnt signalling, Y654 phosphorylation of β-catenin has been implicated in the acquisition of an invasive phenotype of tumour cells.6 According to this model, colorectal tumour cells exposed to high levels of growth factor signalling coming from the tumour stroma induce tyrosine phosphorylation of β-catenin, resulting in reduced cell adhesion and increased nuclear signalling, leading to an epithelial to mesenchymal transition (EMT).6 Our results clearly show, however, that Y654 phosphorylation by itself is not sufficient to induce a migratory and invasive phenotype. Epithelial tissues were properly formed in β-cateninE654/E654 embryos and also in the intestinal tumours we did not observe enhanced invasiveness of tumour cells. These observations do not exclude that Y654 phosphorylation contributes to the induction of EMT, but it will most likely require the simultaneous activation of other EMT inducers and the acquisition of additional mutations, for example in KRAS and/or P53, to uncover its invasion-inducing potential fully.
Based on our observations, we propose the following model (figure 7). In cells that are exposed to extracellular Wnt ligands or have generated a defect in the β-catenin degradation system, β-catenin degradation is inhibited. Activation of RTK results in phosphorylation of β-catenin at Y654 introducing a conformational change affecting protein–protein interactions. This reduces its affinity for the adherens complex, while it is simultaneously more accessible for additional phosphorylation, such as phosphorylation of S675 by PKA. Next, phosphorylation of both Y654 and S675 increases the recruitment of proteins involved in the transcription machinery such as TCF4, CBP and TBP,10 29 30 resulting in increased Wnt signalling. Overall, our data provide in-vivo evidence that in addition to genetic alterations in the intestinal epithelium, the tumour stroma substantially contributes to the development of intestinal cancer through Y654 phosphorylation of β-catenin. Our results suggest that treatment targeted at the tumour stroma, tyrosine kinases and PKA may serve an efficient therapy strategy for patients with intestinal or other cancers involving active Wnt signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of β-catenin Y654 phosphorylation contributing to Wnt signalling. Upon activation of receptor tyrosine kinases (RTK), by overexpression, activating mutations or (stromal) growth factors, β-catenin is phosphorylated at Y654. This introduces a negative charge that evokes a conformational change in the C-terminus of β-catenin, affecting protein–protein interactions. One of the affected interactions is with cadherins for which the affinity is reduced, increasing the amount of free β-catenin. Due to the open structure at the C-terminus, S675 is more accessible for phosphorylation by protein kinase A (PKA). The resulting C-terminal phosphorylated β-catenin enhances the recruitment of proteins of the transcription machinery, such as TCF4, TBP and CBP. Ultimately, this results in increased Wnt signalling contributing to intestinal tumorigenesis.
Acknowledgments
The authors are grateful to John Kong-a-San for ES cell injections and to the animal caretakers.
References
Supplementary materials
Online only appendix
Files in this Data Supplement:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Funding This study was financially supported by grants from the Netherlands Organisation for Scientific Research (NWO/Vidi 917.56.353; NWO/Vici 016.036.636) and the Dutch Cancer Society (DDHK20053299).
Competing interests None.
Ethics approval All animal experiments were approved by the Institute's Animal Ethics Committee and were carried out in accordance with Dutch and international legislation.
Provenance and peer review Not commissioned; externally peer reviewed.