Article Text

Abstract
Background: Serrated adenomas (SAs) of the colorectum combine architectural features of hyperplastic polyps and cytological features of classical adenomas. Molecular studies comparing SAs and classical adenomas suggest that each may be a distinct entity; in particular, it has been proposed that microsatellite instability (MSI) distinguishes SAs from classical adenomas and that SAs and the colorectal cancers arising from them develop along a pathway driven by low level microsatellite instability (MSI-L).
Aims: To define the molecular characteristics of SAs of the colorectum.
Materials and methods: We analysed 39 SAs from 27 patients, including eight SAs from patients with familial adenomatous polyposis (FAP). We screened these polyps for selected molecular changes, including loss of heterozygosity (LOH) close to APC (5q21) and CRAC1 (15q13-q22), MSI, and mutations of K-ras, APC, p53, and β-catenin. Expression patterns of β-catenin, p53, MLH1, MSH2, E-cadherin, and O6-methylguanine DNA methyltransferase (MGMT) were assessed by immunohistochemistry. Comparative genomic hybridisation was performed on several polyps.
Results: MSI was rare (<5% cases) and there was no loss of expression of mismatch repair proteins. Wnt pathway abnormalities (APC mutation/LOH, β-catenin mutation/nuclear expression) occurred in 11 SAs, including 6/31 (19%) non-FAP tumours. CRAC1 LOH occurred in 23% of tumours. K-ras mutations and p53 mutations/overexpression were found in 15% and 8% of SAs, respectively. Loss of MGMT expression occurred in 18% of polyps and showed a borderline association with K-ras mutations. Aberrant E-cadherin expression was found in seven polyps. Comparative genomic hybridisation detected no gains or deletions of chromosomal material.
Conclusions: The serrated pathway of colorectal tumorigenesis appears to be heterogeneous. In common with classical adenomas, some SAs develop along pathways involving changes in APC/β-catenin. SAs rarely show MSI or any evidence of chromosomal-scale genetic instability. K-ras mutations may however be less common in SAs than in classical adenomas. Some SAs may harbour changes in the CRAC1 gene. Changes in known genes do not account for the growth of the majority of SAs.
- serrated adenoma
- microsatellite instability
- loss of heterozygosity
- immunohistochemistry
- CGH, comparative genomic hybridisation
- LOH, loss of heterozygosity
- MSI, microsatellite instability
- MSI-L, low level microsatellite instability
- MSI-H, high level microsatellite instability
- MSS, microsatellite stable
- MGMT, O6-methylguanine DNA methyltransferase
- SSCP, single strand conformational polymorphism
- FAP, familial adenomatous polyposis
- SA, serrated adenoma
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- CGH, comparative genomic hybridisation
- LOH, loss of heterozygosity
- MSI, microsatellite instability
- MSI-L, low level microsatellite instability
- MSI-H, high level microsatellite instability
- MSS, microsatellite stable
- MGMT, O6-methylguanine DNA methyltransferase
- SSCP, single strand conformational polymorphism
- FAP, familial adenomatous polyposis
- SA, serrated adenoma
- PCR, polymerase chain reaction
Serrated adenomas (SAs), first described as a distinct entity by Longacre and Fenoglio-Preiser in 1990,1 combine the architectural features of hyperplastic polyps (glandular lumen serration) and the cytological features of adenomas. Their prevalence at colonoscopy is 1–7%, with the majority (54%) being found in the rectum and sigmoid colon.1,2
The relationship between SAs, hyperplastic (metaplastic) polyps, and classical (tubular, tubulovillous, and villous) adenomas is not clear. Some evidence connects the classical and serrated pathways: for example, SAs can occur in familial adenomatous polyposis (FAP), individuals may have synchronous or metachronous adenomas of both classical and serrated types, and tumours may contain both classical and serrated components.1 Molecular studies comparing SAs and tubular adenomas have however suggested that they are distinct entities, each with their own characteristic spectra of molecular changes. Two studies have shown SAs to have a low K-ras mutation rate compared with tubular adenomas,3,4 although this has not been confirmed in two other studies.5,6 Microsatellite instability (MSI) is another characteristic of SAs that appears to distinguish them from tubular adenomas. In one series, 53% of SAs showed MSI, mostly of the low level (MSI-L) type.7 Very few sporadic classical adenomas show MSI.8 p53 mutations, which are considered a late event in the adenoma-carcinoma pathway, have been documented as being as frequent as 47% in SAs.5 The high p53 mutation rate and detection of significant dysplasia in 13–37% of SAs,9 10–15% having been shown to contain carcinomas in situ,3 has led some to believe that SAs have a greater malignant potential than classical adenomas. However, not all molecular studies have shown consistent findings and earlier studies may have been biased towards larger tumours which were more likely to show dysplasia.10
Hyperplastic polyps have been considered non-neoplastic and have not been thought to be associated with malignancy, except in the hyperplastic polyposis syndrome. This is a rare disease in which large hyperplastic polyps (>1 cm) are distributed throughout the colon, and synchronous dysplasia is common.11 Many of the polyps in this disease might therefore now be classified as SAs, and the condition provides evidence for a hyperplastic polyp-SA sequence. Sporadic hyperplastic polyps are one of the commonest lesions in the large bowel. They are smaller than SAs—two thirds are 5 mm or less in diameter—and they are rarely greater then 1 cm.9,10,12–,14 Recently, sporadic hyperplastic polyps have been shown to exhibit some molecular changes, including K-ras mutation (13–75%)4,5 and MSI-L (30%).7 Hyperplastic polyps also have similar apomucin changes to SAs, expressing gastric mucin M1, MUC2, and reduced or absent MUC4.15 Many currently believe that sporadic hyperplastic polyps should be regarded as benign neoplastic lesions that may have a low malignant potential.
It remains unclear therefore whether or not SAs, and colorectal cancers arising from them, follow a different pathway of tumorigenesis from most colorectal tumours. Jass and colleagues6 proposed that the serrated pathway is driven by MSI: the MSI-L hyperplastic polyp is the precursor lesion which develops into MSI-L colorectal cancer through the intermediate step of the MSI-L SA. Jass and colleagues have also found that K-ras mutations are more frequent in certain MSI-L cancers and that this may be due to silencing of O6-methylguanine DNA methyltransferase (MGMT) expression by methylation.16
In this study, we have documented selected molecular changes in a series of 39 SAs from the UK. We have assessed MSI, loss of heterozygosity (LOH) close to APC (5q21) and on 15q13-q22 (the site of CRAC1, a novel colorectal adenoma and carcinoma susceptibility gene17), and mutations of K-ras, APC, p53, and β-catenin. Expression patterns of β-catenin, p53, MLH1, MSH2, E-cadherin, and MGMT have been assessed using immunohistochemistry. We have also performed comparative genomic hybridisation (CGH) on selected tumours in order to search for additional genetic changes in SAs and to test for the presence of chromosomal instability.
MATERIALS AND METHODS
SA samples were collected from St Marks Hospital. Haematoxylin-eosin stained slides of each case were reviewed by two histopathologists (AC and ICT) independently, to confirm the diagnosis and provide a consistent histological classification. All tumour material was archival, paraffin embedded tissue. Five sections (10 μm) were cut from each tumour and stained with toluidine blue. Each section was microdissected by hand to remove the normal tissue, which was used as a source of constitutional DNA. Wherever possible, DNA derived from blood was also used as a source of constitutional DNA. For mixed tumours of any type, the serrated adenomatous component was microdissected, but due to the small size of the tumours, it was impossible to exclude contamination of the SA by either the adjacent hyperplastic or tubulovillous component. Extraction of DNA from the tumour and normal tissue was performed using the Qiagen (Hilden, Germany) tissue extraction kit and from blood using standard methods.
For the LOH studies, polymorphic microsatellite markers in the region of the APC gene (D5S82, D5S656, D5S489) and the CRAC1 gene (D15S118, D15S1016, D15S1006, D15S1032) were chosen from public databases (http://www.genethon.fr/ and http://www.chlc.org/). MSI was assessed using the chromosome 5 and 15 markers and also D2S123 (dinucleotide), D11S29 (dinucleotide), BAT26 (mononucleotide), MYCL (mixed repeat), D15S659 (tetranucleotide), and D15S644 (tetranucleotide). Microsatellite loci were amplified in the polymerase chain reaction (PCR) in a 25 μl volume, with one oligonucleotide fluorescence labelled. The PCR reaction typically contained 20–100 ng DNA, 50 mM KCl, 0.5–2.5 mM MgCl2, 10 mM Tris-HCl, 0.1% Triton, 2.5 μg bovine serum albumin, 0.2 mM of each dNTPs, 10 pmol of each oligonucleotide, and 1.25U Taq DNA polymerase (Promega, Madison, Wisconsin, USA). The PCR reaction consisted of an initial step of 94°C for four minutes, then 40 cycles of one minute at 94°C, one minute at the appropriate annealing temperature, and one minute at 72°C in a PTC-225 Peltier thermal cycler (MJ Research, Waltham, Massachusetts, USA). Microsatellites were analysed for LOH using the Genotyper programme (ABI, Foster City, California, USA). LOH at each marker locus was considered to be present if the area under one allelic peak in the tumour was less than 0.5× or greater than 2× that of the other allele, after correcting for the relative allelic areas using the constitutional DNA. Tumours showing novel bands were classed as MSI+. All PCR reactions were repeated on the same sample and only changes consistent between the duplicated reactions were scored as abnormalities.
Codons 12 and 13 of K-ras were amplified in the PCR using previously published primers.5 The amplified product was then directly sequenced in forward and reverse directions, using the ABI sequencer and standard protocols.
Exons 5–8 of p53 were amplified in the PCR using previously published primers5,18 and subjected to single stranded conformational polymorphism (SSCP) analysis. The amplified fragments (10 μl) were mixed with 10 μl of loading buffer (98% deionised formamide, 10 mM EDTA, 0.025% xylene cyanole FF, and bromophenol blue mixed in a ratio of 5:7 with 0.1% sodium dodecyl sulphate, 10 mM EDTA), heat denatured at 95°C for 10 minutes and loaded onto an 8% non-denaturing polyacrylamide midi-gel containing 10% glycerol. Electrophoresis was carried out at a constant power of 6 W for 13–15 hours at room temperature. The gels were silver stained using standard protocols. PCR and direct sequencing were performed on those samples which showed a mobility shift or extra banding on SSCP gels.
Regions G, H, and I of exon 15 of the APC gene (corresponding to the mutation cluster region) were screened for mutations using primers designed for archival material which amplify 11 overlapping short fragments (details available from the authors). Each sample was screened for mutations using SSCP as above. For those samples which showed a mobility shift or extra banding on SSCP gels, the DNA fragment was reamplified and sequenced either directly or after subcloning into plasmid pGEM-T-Easy.
Exon 3 of β-catenin was amplified in the PCR using the following primers: sense 5`-ATTTGATGGAGTTGGACATGGC-3`, antisense 5`-CCAGCTACTTGTTCTTGAGTGAAGG-3`. SSCP analysis was performed using the Phast System (Pharmacia, Uppsala, Sweden). The PCR products were denatured at 95°C for five minutes and subjected to electrophoresis on a 20% precast polyacrylamide gel. DNA was detected by silver staining of gels using the standard Phast System protocol. PCR and direct sequencing were performed on those samples which showed a mobility shift or extra banding on SSCP gels.
For immunohistochemistry, serial sections (4 μm) were deparaffinised, rehydrated, and incubated with the different mouse monoclonal antibodies for one hour. Antigen retrieval was used for β-catenin, MLH1, MSH2, E-cadherin, and MGMT (pressure cooking two minutes at full pressure in 3 litres of 0.01 M citrate buffer). Dilutions were 1 in 70 for p53 (Oncogene Science), 1 in 800 for β-catenin (Transduction Laboratories, San Diego, California, USA), 1 in 100 for MLH1 (Pharmingen, San Diego, California, USA), 1 in 100 for MSH2 (Calbiochem, San Diego, California, USA), 1 in 20 for E-cadherin (ICRF), and 1 in 50 for MGMT (Neomarkers, Lab Vision Corporation, Fremont, California, USA). Biotinylated rabbit antimouse immunoglobulin diluted 1 in 400 in 3% normal human serum was used as the secondary antibody. After washing, the slides were incubated in peroxidase conjugated streptavidin-biotin complex (Dako, Copenhagen, Denmark) for 30 minutes. The peroxidase was demonstrated by incubating in diaminobenzidine solution for five minutes. After rinsing in water, nuclei were lightly counterstained with haematoxylin for two minutes and coverslipped. The sections were then scored by a histopathologist (AH or MJA) for presence, location, and intensity of staining (3, strong; 2, moderate; 1, weak; 0, no staining).
CGH was performed on four tumours. Tumour and control DNAs were labelled by nick translation and hybridised to normal female metaphase spreads in the presence of human CotI DNA using standard protocols.19 Metaphase chromosome preparations were captured using Smartcapture software (Vysis). Five to 10 representative images were analysed using Quips CGH software (Vysis).
RESULTS
In this series of 39 SAs, the majority (25/39, 64%) showed mild dysplasia (table 1⇓, fig 1⇓); 11 tumours (28%) showed moderate dysplasia and three (8%) showed severe dysplasia. One tumour (No 30) contained both SA and carcinoma. There were four mixed tubulovillous/SAs (Nos 2, 19, 24, 36) and three mixed hyperplastic polyps/SAs (Nos 1, 14, 25). The majority of cases (79%) were from the distal colon or rectum and the average maximum diameter was 0.9 cm. Eight tumours came from five patients with FAP. A further 14 patients were known to have had a history of sporadic classical adenomas, and of those, seven had also had previous hyperplastic polyps.
Summary of clinicopathological data
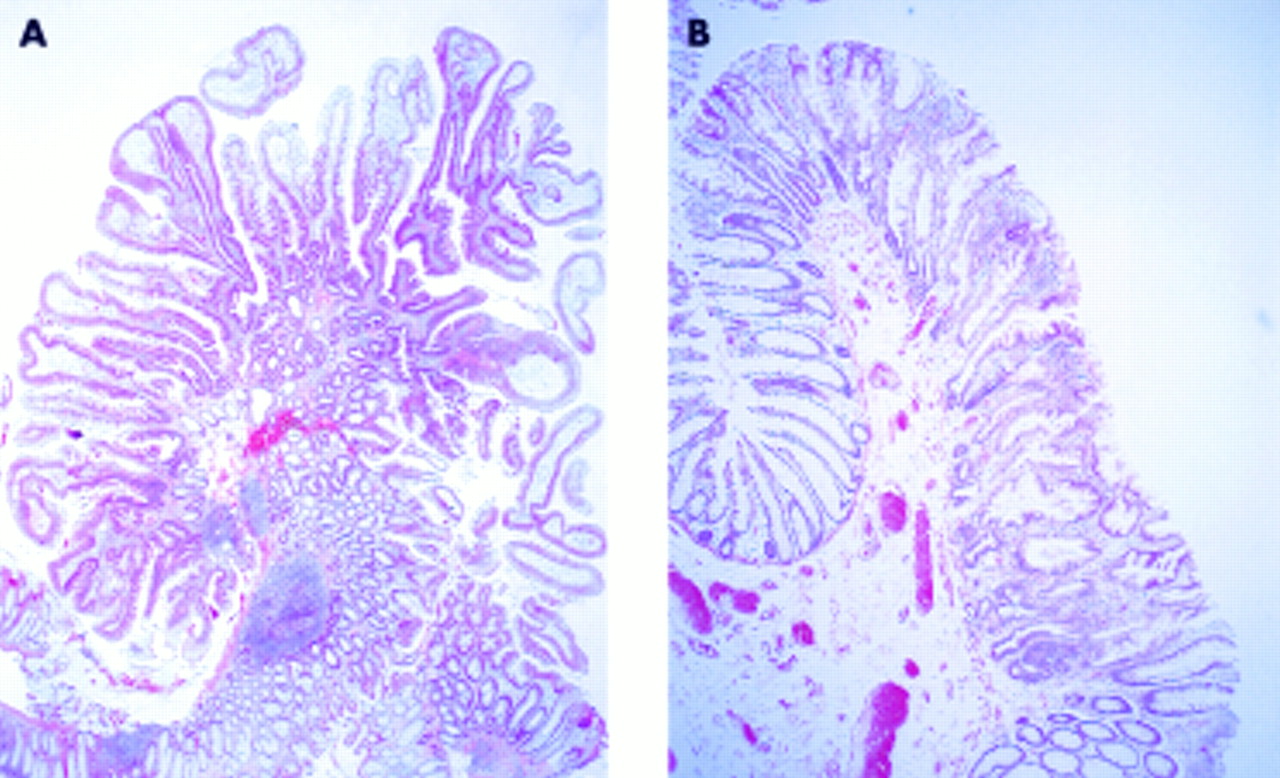
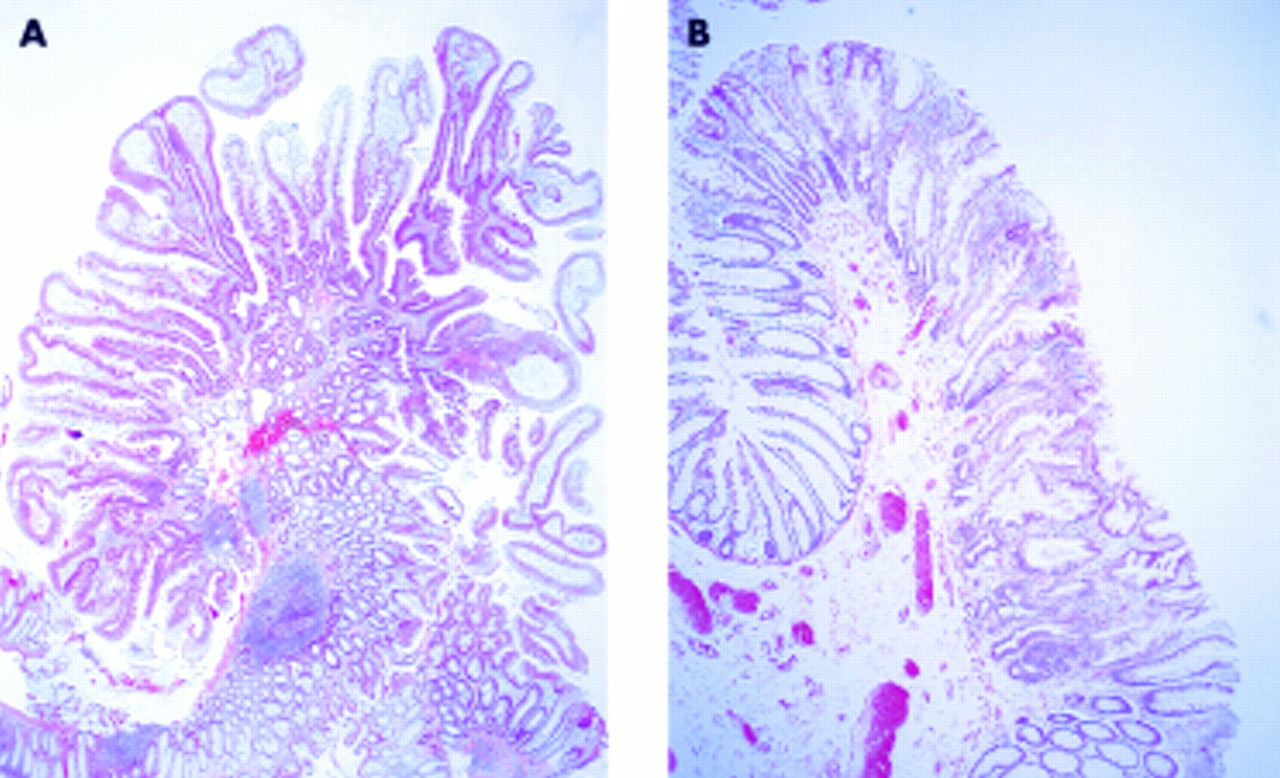
(A) Case No 13—serrated adenoma with mild dysplasia. (B) Case No 6—serrated adenoma with mild dysplasia.
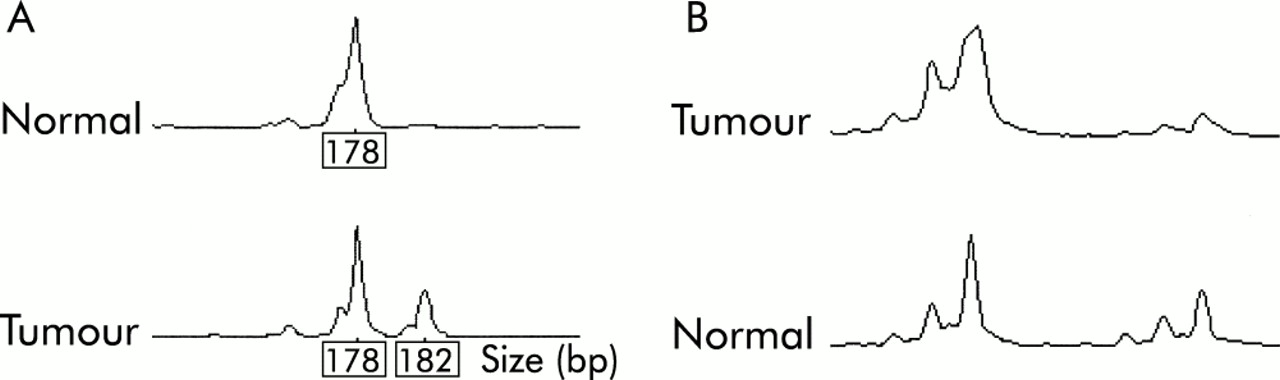
Two tumours (5%) showed MSI-L (fig 2A⇓), one at MYCL only (No 37) and one at D15S118 only (No 3). In both cases, a single repeat unit bandshift was observed. None of the tumours showed MSI at D2S123, D11S29, BAT26, or at the other markers used. No loss or other abnormality of MLH1 or MSH2 staining was found in any of the tumours.
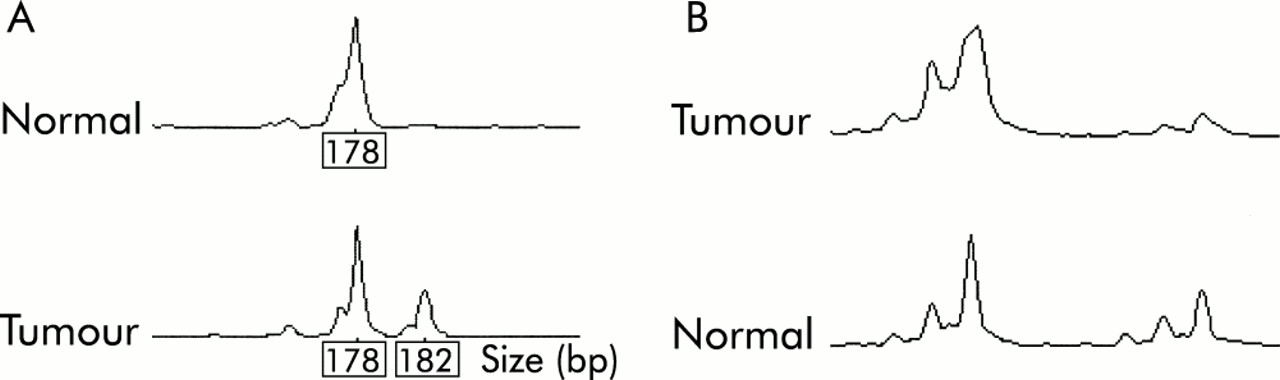
(A) Low level microsatellite instability in sample No 37 using marker MYCL. (B) Loss of heterozygosity at D5S656 in sample No 7.
Thirty samples were available for LOH analysis, the remaining nine samples having no source of paired normal DNA. Two tumours (Nos 7, 32) showed LOH of at least one marker in the region of APC (fig 2B⇑). Neither of these tumours was from an FAP patient and in neither was a truncating APC mutation subsequently found. Somatic mutations in the mutation cluster region of the APC gene (fig 3A⇓, table 2⇓) were found in five tumours (13%); one of these tumours was a mixed tubulovillous/SA but the rest were pure SAs. Four of the five tumours with somatic APC mutations were from FAP patients. One codon 33 β-catenin mutation was found in a tumour (No 31) with severe dysplasia and abnormal (cytoplasmic) β-catenin expression on immunohistochemistry (fig 3B⇓). Five tumours showed nuclear β-catenin staining; in three cases this was focal and in the others widespread (table 2⇓). Two of the polyps (Nos 4 and 13) with nuclear β-catenin staining came from FAP patients, and another (No 7) showed LOH at APC.
Summary of molecular data

{kind=link}
{kind=link}
{kind=link}
(A) 5 base pair deletion at codon 1309 of the APC gene in sample No 15. (B) β-catenin mutation codon 33 (TCT→TGT) in sample No 31.
Seven of 30 tumours (23%) showed LOH on 15q in the region of the CRAC1 gene (table 2⇑), three at D15S016, two at D15S644, one at D15S1006, and one at D15S118. Six tumours (15%) showed mutation of K-ras, five at codon 12 and one at codon 13 (table 2⇑).
Two tumours (5%) were found to have p53 mutations, both in exon 5: one mutation occurred in the sample with associated carcinoma (No 30) which also had nuclear p53 staining on immunohistochemistry and the other mutation occurred in the mixed tubulovillous/SA (No 2) which had normal p53 immunohistochemistry. One other tumour (No 15) showed abnormal expression of p53 but had no detectable p53 mutation.
Loss of expression of MGMT occurred in seven tumours (table 2⇑), of which three had mutations in K-ras. There was a borderline association between loss of MGMT expression and K-ras mutation (p=0.06, Fisher's exact test). Three tumours showed reduced membrane expression of E-cadherin and four strong cytoplasmic staining with E-cadherin (table 2⇑).
Comparative genomic hybridisation was performed on samples Nos 15, 30, 38, and 39. No chromosomal-scale imbalances were found in any tumour.
DISCUSSION
The serrated pathway of colorectal tumorigenesis appears to be heterogeneous, with both similarities to and differences from the “classical” pathway. The first point of similarity is the low level of MSI in both types of lesion. In our study, only two SAs showed MSI-L despite our use of multiple markers of different types, and no tumour showed loss of mismatch repair proteins. The reasons for these difference between our study and others7 are unclear because similar microsatellite markers were used to assess the tumours. One reason may be differences in the tumours studied. Previous series have shown high level microsatellite instability (MSI-H) to be infrequent in sporadic serrated polyps (in contrast with serrated polyps from patients with hyperplastic polyposis20) and where MSI-H does occur it appears more frequently in proximal tumours. MSI-H was found in 17% (5/29) of cases by Jass and colleagues.20 These polyps were proximal and mixed serrated/hyperplastic. Hiyama and colleagues5 found only 1/19 SAs to be MSI-H. In a recent study by Makinnen et al, eight of the 27 carcinomas associated with SAs were analysed for MSI and in three cases (37.5%), all proximal, MSI was found.21 We have also found that repeating the PCR amplification where the reaction contains small amounts of target DNA of potentially low quality can be important in determining true MSI. In the first PCR, 12 samples appeared to show MSI (four at D2S123, two at D11S29, one at D15S118, one at D15S644, and four at MYCL). All of these showed a single extra discrete band in tumour DNA. No sample showed a marked alteration in repeat length appearing as a ladder. On repeating the PCR for these samples, apart from the two tumours with instability of MYCL and D15S118, none of these additional bands was present, suggesting that they were artefacts. Detection of MSI can therefore be problematic in DNA extracted from paraffin embedded tissue and its prevalence may have been overestimated. MYCL may however be a relatively sensitive and specific marker for MSI, as Jass et al have reported.22
In common with the majority of classical adenomas, a minority of SAs in our series developed along pathways involving changes in APC/β-catenin. In our series, eight SAs came from patients with FAP and six other SAs had alterations in APC and/or β-catenin (table 2⇑). This association has been noted previously,1 but despite this, many studies on SAs have not looked at APC directly but have used 5q21 LOH or nuclear β-catenin staining to infer APC mutation. Our study shows that APC mutations in SAs can occur in the absence of LOH and/or nuclear β-catenin staining, as previous studies have shown for classical adenomas.23,24 All studies that have looked for APC mutations in SAs outside FAP have shown a low rate of detection4,25 relative to the 30–64% reported in classical adenomas (for example, see Miyaki and colleagues24 and Morris and colleagues26). However, plentiful high quality DNA has been used in many of the studies of classical adenomas in contrast with the much more limited analysis which is possible using the archival material analysed here. We cannot therefore exclude the possibility that APC mutations are involved in the pathogenesis of a much greater proportion of SAs than we have found.
p53 mutations or aberrant expression occurred in fewer than 10% of our SAs compared with 47% in one previous study.5 This difference is likely to reflect the fact that the majority of tumours in our series showed only mild dysplasia. p53 mutations are also uncommon in early adenomas of classical morphology. A further point of similarity between SAs and classical adenomas is the absence of chromosomal-scale genetic instability, as assessed using CGH (for example, see Barletta and colleagues27).
Our data also suggest some differences between SAs and classical adenomas. The finding of LOH at CRAC1 in 23% of SAs is intriguing given the presence of both serrated and classical adenomas in families with germline CRAC1 mutations.17 However, these data require confirmation once the position of CRAC1 has been refined and some caution also as individual markers only were lost. Our preliminary data suggest that LOH at CRAC1 occurs in fewer than 15% of classical adenomas (Jaeger et al, unpublished observations).
K-ras mutations occurred in about 15% of our tumours. The detection method for K-ras mutations was designed to be sensitive. There were few K-ras mutations in this study compared with published series of sporadic classical adenomas (44%), suggesting that this may be a true difference between the two types of adenoma.5,26 The fact that K-ras mutations were more common in tumours with loss of MGMT expression supports some of the findings of Whitehall and colleagues,16 but unlike their study, MGMT loss did not occur in our tumours which showed MSI-L.
Our study suggests that APC and other Wnt pathway abnormalities may be important in the pathogenesis of a subset of SAs of the colorectum as well as in classical adenomas. The CRAC1 gene may also be involved in serrated tumorigenesis. Contrary to previous data, MSI was rare in sporadic and FAP associated SAs and p53 abnormalities were uncommon, at least in the absence of moderate or severe dysplasia. In accordance with previous studies, we found that K-ras mutations were less frequent in SAs than classical adenomas. The differences between our study and previous ones may reflect different populations—many of the molecular studies on SAs have been performed on Japanese cases5,7,10,28—and the increasing recognition by histopathologists of more subtle manifestations of the serrated phenotype.
Acknowledgments
We are grateful to the Equipment Park, ICRF, for gel running. ES is supported by the Special Trustees of Guy's and St Thomas' Hospital and the Imperial Cancer Research Fund.