Article Text

Abstract
Objective Insulinomas and non-functional pancreatic neuroendocrine tumours (NF-PanNETs) have distinctive clinical presentations but share similar pathological features. Their genetic bases have not been comprehensively compared. Herein, we used whole-genome/whole-exome sequencing (WGS/WES) to identify genetic differences between insulinomas and NF-PanNETs.
Design The mutational profiles and copy-number variation (CNV) patterns of 211 PanNETs, including 84 insulinomas and 127 NF-PanNETs, were obtained from WGS/WES data provided by Peking Union Medical College Hospital and the International Cancer Genome Consortium. Insulinoma RNA sequencing and immunohistochemistry data were assayed.
Results PanNETs were categorised based on CNV patterns: amplification, copy neutral and deletion. Insulinomas had CNV amplifications and copy neutral and lacked CNV deletions. CNV-neutral insulinomas exhibited an elevated rate of YY1 mutations. In contrast, NF-PanNETs had all three CNV patterns, and NF-PanNETs with CNV deletions had a high rate of loss-of-function mutations of tumour suppressor genes. NF-PanNETs with CNV alterations (amplification and deletion) had an elevated risk of relapse, and additional DAXX/ATRX mutations could predict an increased relapse risk in the first 2-year period.
Conclusion These WGS/WES data allowed a comprehensive assessment of genetic differences between insulinomas and NF-PanNETs, reclassifying these tumours into novel molecular subtypes. We also proposed a novel relapse risk stratification system using CNV patterns and DAXX/ATRX mutations.
- pancreas
- neuroendocrine tumours
- gene mutation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The two most common types of pancreatic neuroendocrine tumours (PanNETs), insulinomas and non-functional PanNETs (NF-PanNETs), exhibit different clinical presentations, different metastatic rates and prognoses.
Sequencing studies have revealed distinctive significantly mutated genes (SMGs) in insulinomas and NF-PanNETs. In particular, YY1 is the only SMG identified by whole-exome sequencing (WES) of insulinomas, of which YY1-mutated insulinomas account for only a minor proportion.
DAXX/ATRX mutations have been proposed to be a prognostic marker for NF-PanNETs.
What are the new findings?
The integrated analyses of whole-genome sequencing/WES data revealed distinctive copy-number variation (CNV) and single-nucleotide variant patterns and reclassified insulinomas and NF-PanNETs into five molecular subtypes.
YY1-mutated insulinomas tended to belong to the copy neutral subtype. Insulinomas without YY1 mutations had a high prevalence of CNV amplification. In particular, chromosome 7 amplifications were identified as a common early event for insulinomas with CNV amplifications.
In NF-PanNETs, the presence of CNV alterations (amplification and deletion) was identified as a prognostic marker for high relapse risk and was correlated with advancement of clinicopathological parameters. In NF-PanNETs with CNV amplification/deletion, if DAXX/ATRX genes were also mutated, the relapse risk was elevated in the first 2 years after surgery.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
In insulinomas, the CNV pattern serves as a useful classifier for different tumourigenesis mechanisms. In particular, CNV neutral insulinomas are mainly driven by YY1 mutations.
We propose a relapse risk stratification system for PanNETs. The relapse risk could be categorizedcategorised by CNV pattern. For those with high relapse risk, DAXX/ATRX mutations were correlated with early relapse.
Introduction
The incidence and prevalence of pancreatic neuroendocrine tumours (PanNETs) have increased. Specifically, the incidence increased substantially from 0.32/100 000 persons during the Surveillance, Epidemiology and End Results Program (SEER17) period (2000–2004)1 to 0.48/100 000 persons in the SEER18 period (2000–2012) in the USA.2 PanNETs include both functional PanNETs and non-functional PanNETs (NF-PanNETs), depending on whether functional hormonal hypersecretion syndrome is present.3 4 The most common type of functional PanNETs is insulinoma.
Pathological similarities have been found between insulinomas and NF-PanNETs, which are stained positively for neuroendocrine markers. However, insulinomas have less malignant clinical characteristics than NF-PanNETs.5 6 Although both insulinomas and NF-PanNETs originate from pancreatic endocrine cells, miRNA and mRNA transcriptome studies showed that insulinomas originate from mature β-cells, whereas NF-PanNETs can originate either from β-cell precursors or through the dedifferentiation of insulinomas via epithelial–mesenchymal transition.7
Whether insulinomas and NF-PanNETs share common genetic bases during tumourigenesis remains controversial. Previous studies have identified recurrent YY1 T372R mutations in approximately 15%–32% of patients with insulinomas8–10; but similar mutations have not been identified in patients with NF-PanNETs.11 12 In contrast, MEN1, DAXX/ATRX and mTOR pathway genes (including PTEN, TSC1, TSC2, PIK3CA and DEPDC5) are found to be frequently mutated in approximately 55%–65% of patients with NF-PanNETs,12 13 but the same mutations have rarely been observed in insulinomas.8–10 These findings indicated that insulinomas and NF-PanNETs had discrete genetic bases with regard to somatic mutations. However, significant proportions of PanNETs, particularly insulinomas, lack the aforementioned mutations. Moreover, previous studies on insulinomas were mainly confined to whole-exome sequencing (WES). Here, we conducted whole-genome sequencing (WGS)/WES of PanNET patients from Peking Union Medical College Hospital (PUMCH) and used curated WGS data from the International Cancer Genome Consortium (ICGC) project.12 RNA sequencing of PUMCH insulinomas was also performed. We aimed to address genetic differences and prognostic significance through comprehensive comparisons of sequencing data from PanNET patients.
Materials and methods
Patient inclusion and exclusion criteria
The inclusion criteria were as follows: (1) clinical diagnoses of insulinomas or NF-PanNETs according to published criteria5 and (2) pathological diagnoses of G1, G2 or well-differentiated G3. The exclusion criteria were as follows: (1) clinical diagnoses of functional PanNETs other than insulinomas and (2) histological diagnoses of poorly differentiated G3 PanNETs or PanNETs combined with pancreatic adenocarcinomas. Fresh frozen tumours and either paired normal tissues or peripheral blood samples were obtained from Clinical Biobank, PUMCH and formalin-fixed, paraffin-embedded (FFPE) tissues were obtained from the Department of Pathology, PUMCH. Identifiable patient information (name and ID) was deidentified and randomly assigned a study ID number to prevent tracking of the data back to individual patients. We obtained clinical information from the published ICGC dataset.12 The American Joint Committee on Cancer staging system was used for the PanNETs.14
WGS, WES and RNA sequencing
WGS libraries were constructed from fresh frozen tissues or peripheral blood samples, and WES libraries were constructed from both fresh frozen tissues, peripheral blood samples and FFPE tissues as described previously.15 The constructed libraries were sequenced using BGISEQ-500 sequencing platforms as paired-end 100 bp reads. High-quality sequencing reads were processed using DRAGEN software, including alignment to the human reference genome (hg19), sorting and duplicate marking. For RNA sequencing, complementary DNA (cDNA) was prepared as previously described.15 Then, the cDNA libraries were constructed for fresh frozen tumour tissues and sequenced using BGISEQ-500 platform. The gene expression levels were calculated according to the fragments per kilobase of exons per million reads method using RSEM.16
Analysis of ICGC datasets
ICGC datasets were obtained from the European Genome-Phenome Archive (accession number EGAS00001001732). All downloaded bam files were realigned to hg19 reference sequence by DRAGEN, and then new bam files and PUMCH datasets were combined for all subsequent analyses.
Mutation analysis
Germline variants were called by DRAGEN software. Somatic mutations were identified by MuTect17 for single nucleotide variants (SNVs) and Platypus18 for short insertions and deletions. All detected mutations were annotated with oncotator.19 To obtain high-quality somatic mutations, we used the Panel of Normal filter of MuTect, which was constructed from all normal samples (fresh frozen paired normal tissues and peripheral blood samples), and candidates that presented in two or more normal samples were rejected. The pathogenicity of germline variants was predicted according to criteria described in the American College of Medical Genetics and Genomics guidelines.20 In brief, we selected pathogenic variants based on whether the variants were predicted to disrupt gene function, their frequency in population sequencing databases such as the Exome Aggregation Consortium and 1000 Genomes, and their clinical significance according to ClinVar.21 For somatic mutations, significantly mutated genes (SMGs) were identified with IntOGen.22
Copy-number variations and structural variations
The integer copy numbers were estimated with FACETS.23 The copy-number variation (CNV) status was determined by the threshold of >2 copies for amplification and <2 copies for deletion. For amplified segments, we used Palimpsest24 to determine the molecular timing of amplification, which was represented as the point mutation time (pmt). When a segment was amplified, the mutations that occurred prior to the amplification event were also amplified, resulting in increased variant allele frequencies. The a segment that is amplified early will have fewer amplified mutations compared with a segment that is amplified late. Therefore, the timing of the amplifications can be estimated by the numbers of amplified and non-amplified mutations. The absence of amplified mutations in a segment (pmt=0) means that the amplification occurred at the very beginning, and the amplification of all mutations in a segment (pmt=100%) indicates that the amplification occurred at a very late stage. Structural variations were detected using SvABA.25
Tissue microarrays, immunohistochemistry and scoring system
The tissue microarrays (TMAs) were constructed with FFPE tissues in accordance with PanNET patients from PUMCH, with one core per patient case. The TMAs were constructed with a diameter of 2 mm for each core and 6 rows and 8 columns of each chip. The phospho-S6 ribosomal protein (Ser235/236) (CST4858, 1:600 dilution with ethylenediaminetetraacetic acid (EDTA) antigen retrieval) was stained according to routine conventional immunohistochemistry (IHC) procedures.26 Cytoplasmic staining intensities of p-S6 were assigned scores of 0 (negative), 1 (weak), 2 (moderate) or 3 (strong). The percentages of tumour cells with positive staining were also recorded. IHC scores ranging from 0 to 300 were calculated by multiplying the staining intensity by the percentage of positive cells, and each TMA core was independently evaluated by at least two pathologists.27
Statistical analysis and data availability
The normality of the data was tested using the Shapiro-Wilk test. The Mann-Whitney U test was used for continuous variables that failed normality test, while Student’s t test was used for continuous variables with normal distributions. Fisher’s exact test and the χ2 test were used for categorical variables. Kaplan-Meier survival and Cox regression were used for survival analyses. All statistical analyses were performed using R.28 Raw sequencing data have been deposited to the China National Genebank Nucleotide Sequence Archive with accession CNP0000383 (https://db.cngb.org/cnsa/).
Results
Clinicopathological parameters and sequencing data
A total of 211 PanNETs, including 84 insulinomas and 127 NF-PanNETs, were analysed. PUMCH patients included 76 insulinomas and 44 NF-PanNETs, whereas the ICGC cohort consisted of 8 insulinomas and 83 NF-PanNETs. Compared with NF-PanNETs, insulinomas were associated with an earlier age of onset, smaller tumour size and lower histological grade (see online supplementary table S1). WGS was performed on 62 insulinomas and 15 NF-PanNETs from PUMCH, with an average depth of 75× for tumours and 42× for the paired samples, which was comparable to the depth obtained in the analysis of ICGC patients (average depth of 61× for tumours, 38× for paired samples).12 A significantly lower tumour mutation burden (TMB) was observed in insulinomas than in NF-PanNETs based on WGS (see online supplementary figure S1). As reported previously, structural variations were present in PanNETs.12 We found that there was no difference between insulinomas (mean, 18 per tumour; range 0–114) and NF-PanNETs (mean, 24 per tumour, range 0–252, p=0.924). For validation, we performed WES on 63 insulinomas and 42 NF-PanNETs from PUMCH (average depth of 686× for tumours and 691×for paired samples), of which 49 insulinomas and 13 NF-PanNETs were also included in the PUMCH WGS dataset. The majority (76/84) of insulinomas were obtained from PUMCH, and enucleations were the most prevalent surgical procedure (63/76) for PUMCH insulinomas. Approximately one-third of the NF-PanNETs (44/127) were from PUMCH, and two-thirds (83/127) were from ICGC. PUMCH NF-PanNETs had a significantly earlier age of onset, smaller tumour size and fewer lymph node metastases than ICGC NF-PanNETs. Other clinicopathological parameters, such as gender, histological grade and distant metastasis rate, were comparable between PUMCH and ICGC NF-PanNETs (see online supplementary table S2).
Supplemental material
Differences in SMGs and CNVs between insulinomas and NF-PanNETs
SMGs were identified using IntOGene(q<0.1) from WGS data for insulinomas and NF-PanNETs, respectively. Four SMGs were identified in insulinomas, and seven SMGs were found in NF-PanNETs. Those SMGs were also screened in all 211 tumours. The most frequent SMGs were YY1 (mutation rate: 25%) in 84 insulinomas, with YY1 T372R mutations as the only mutations. In contrast, the most frequent SMGs were MEN1(42%), DAXX (21%), ATRX (13%), PTEN (9%), and SETD2 (5%) in 127 NF-PanNETs, which was consistent with the findings of previous studies.8–10 12 13 All other SMGs were less frequently mutated (mutation rates <5%) (figure 1A,B). The somatic MEN1 mutation rate was different between insulinomas (1.2%) and NF-PanNETs (42%), and this prompted us to screen for germline mutations of MEN1 in all 211 patients. Pathogenic germline MEN1 mutations were found in 1/84 insulinomas and 6/127 NF-PanNETs. Overall, the MEN1 mutation rates were 2% in insulinomas and 45% in NF-PanNETs. For NF-PanNETs, the mutation rates of the SMGs were without significant differences between PUMCH and the ICGC cohorts (see online supplementary table S2).

Differences in SMGs and CNVs between insulinomas and NF-PanNETs. SMGs and mutation rates in (A) 84 insulinomas and (B) 127 NF-PanNETs. NF-PanNET. Landscapes of somatic CNVs in (C) insulinomas (D) and NF-PanNETs; chromosomes and samples are shown on the y-axis and x-axis, respectively. Cluster classification indicated the presence of two subtypes with distinct CNV patterns within the insulinomas and three subtypes within the NF-PanNETs; the subtypes are highlighted with different colours. CNV, copy-number variation; Ins-Amp, insulinoma amplification subtype; Ins-Neutral, insulinoma neutral subtype; NF-Amp, NF-PanNET amplification subtype; NF-Del, NF-PanNET deletion subtype; NF-Neutral, NF-PanNET neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; SMG, significantly mutated gene.
The somatic CNVs were profiled using WGS data, and unsupervised clustering analysis identified three CNV patterns in PanNETs, namely, amplification, copy neutral and deletion. A recent study showed that WGS outperformed single-nucleotide polymorphism (SNP) arrays in CNV detection.29 As CNVs have been identified in PanNETs by SNP arrays,12 we compared the two methods. The CNV deletion subtype mainly corresponded with the ‘recurrent pattern of whole chromosomal loss’ group. The copy neutral subtype mainly corresponded to the ‘limited copy-number events’ group. The amplification subtype mainly corresponded to the ‘aneuploidy’ and ‘polyploidy’ groups, respectively. Here, insulinomas were classified into either insulinoma amplification (Ins-Amp) or insulinoma copy neutral (Ins-Neutral) subtypes. Meanwhile, NF-PanNETs were classified into three subtypes, namely, NF-PanNET amplification (NF-Amp), NF-PanNET copy neutral (NF-Neutral) and NF-PanNET deletion (NF-Del) subtypes (see online supplementary figure S2A,B). To accurately assign WES data to these subtypes, we estimated the cut-off value for each CNV subtype based on the amplification or deletion fractions. The amplification or deletion fraction distributions were calculated for each CNV group. Then, the cut-off values were set at which the probabilities of a sample being assigned to a deletion/amplification group and to a non-deletion/non-amplification group were equal (see online supplementary figure S2C). We subsequently verified these cut-off values using samples sequences by both WGS and WES; in total, 46/49 (93.9%) insulinomas and 12/13 (92.3%) NF-PanNETs were classified as belonging to the same through both an unsupervised clustering analysis of WGS and by a cut-off value analysis of WES data. Combining the WGS and WES data, there were 29 Ins-Neutral and 55 Ins-Amp (figure 1C) as well as 34 NF-Del, 55 NF-Neutral and 38 NF-Amp (figure 1D). Notably, in contrast to almost 30% of the NF-PanNETs that had deletions, no deletions were present in insulinomas.
Distinctive molecular characteristics between insulinomas and NF-PanNETs within the same CNV patterns
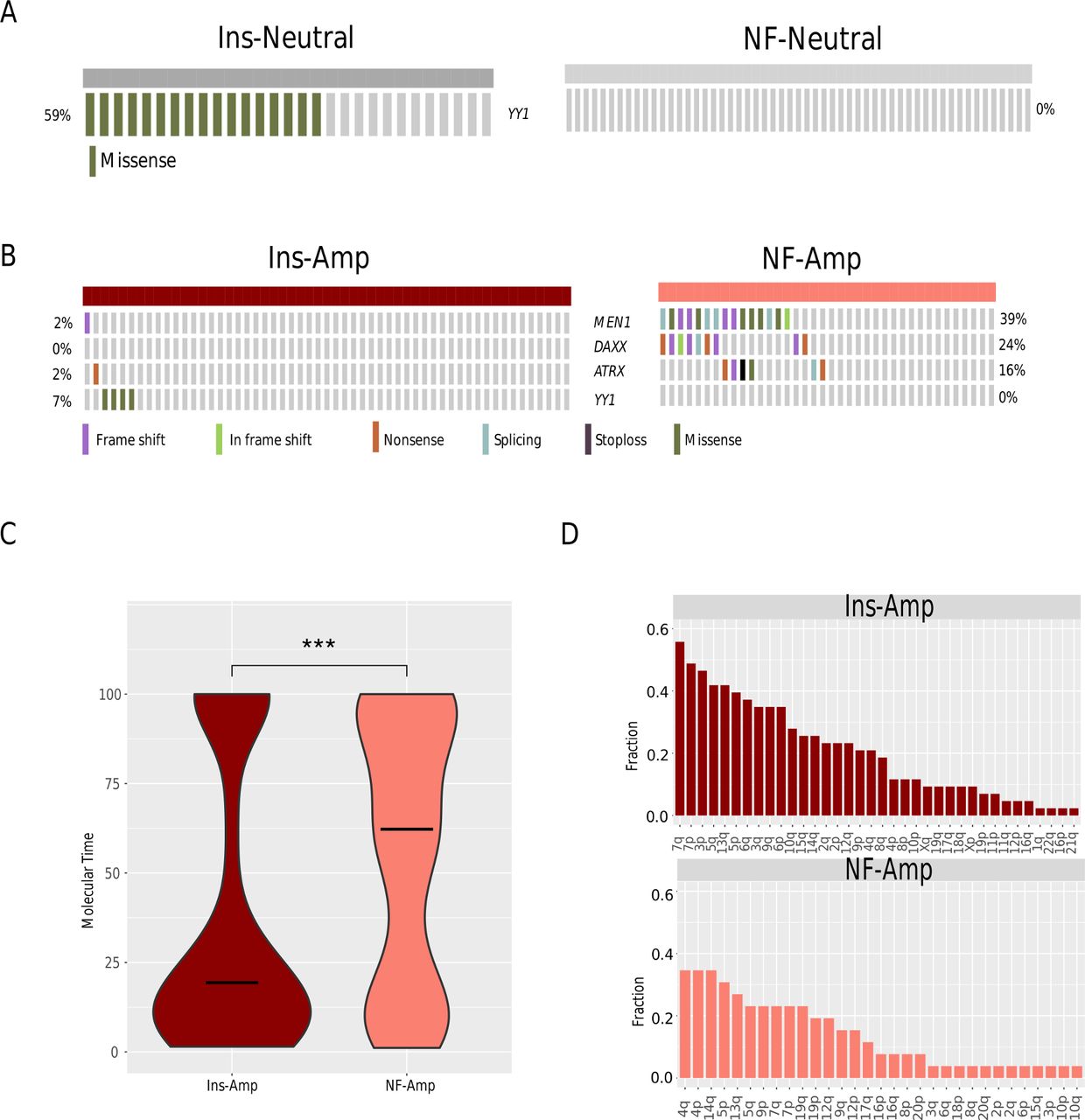
Insulinomas and NF-PanNETs were characterised by different SNVs, even with the same CNV pattern. First, 17/29 Ins-Neutral had YY1 T372R mutations, while no YY1 mutations were detected in NF-Neutral (figure 2A). Meanwhile, no commonly recurring mutations were identified in Ins-Amp. In contrast, MEN1 (15/38) and DAXX/ATRX (15/38) were frequently mutated in the NF-Amp (figure 2B).

Distinctive SNV characteristics within the same CNV patterns. Comparison of frequent mutations between (A) Ins-Neutral and NF-Neutral and between (B) Ins-Amp and NF-Amp. (C) Comparison of the molecular timing of chromosome amplifications between Ins-Amp and NF-Amp. (D) Fractional distributions of chromosomes that were involved in early amplification events (%pmt <20%) in Ins-Amp and NF-Amp tumours. ***P<0.001. CNV, copy-number variation; Ins-Amp, insulinoma amplification subtype; Ins-Neutral, insulinoma neutral subtype; NF-Amp, NF-PanNET amplification subtype; NF-Neutral, NF-PanNET neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; pmt, point mutation time; SNV, single-nucleotide variant.
We further explored whether Ins-Amp and NF-Amp were driven by the same amplification events by estimating the molecular timing using Palimpsest,24 of which the smaller the pmt, the earlier the amplification occurred. Amplifications occurred significantly earlier in Ins-Amp (median timing=19.4% pmt) than in NF-Amp (median timing=62.2% pmt, p<0.001, figure 2C), indicating that early amplification events were more likely to be involved in the tumourigenesis of Ins-Amp than in that of NF-Amp. Furthermore, the common early event (timing <20% pmt) in Ins-Amp was the amplification of chromosomes 7, 3 p, 5q and 13q, while in NF-Amp, it was the amplification of chromosome 4 and 14q (figure 2D). These results indicated that there were different early amplification events in Ins-Amp and NF-Amp.
Five molecular subtypes and prognostic significance
Insulinomas and NF-PanNETs could be categorised into five distinctive molecular subtypes (figure 3A). On the chromosomal alteration axis, the CNV deletion part contained NF-Del; the CNV neutral part contained two subtypes, which were Ins-Neutral and NF-Neutral; the CNV amplification part contained two subtypes, which were Ins-Amp and NF-Amp. The main features of the molecular subtypes were as follows: (1) Ins-Neutral showed a higher YY1 mutation rate; (2) Ins-Amp contained nearly no YY1 mutations and (3) The early amplifications preferred different chromosomes between Ins-Amp and NF-Amp.

Five molecular subtypes and corresponding RFS for insulinomas and NF-PanNETs. (A) The x-axis represents the chromosomal alterations. The y-axis represents the percentage of patients with insulinomas or NF-PanNETs in the current study. (B) RFS among CNV subtypes. CNV, copy-number variation; Ins-Amp, insulinoma amplification subtype; Ins-Neutral, insulinoma neutral subtype; NF-Amp, NF-PanNET amplification subtype; NF-Del, NF-PanNET deletion subtype; NF-Neutral, NF-PanNET neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; RFS, relapse-free survival.
We further analysed relapse-free survival (RFS) for these five molecular subtypes. Ins-Amp and Ins-Neutral had nearly no relapse events. NF-Neutral had relapse events, but its RFS was significantly better than NF-Amp (p=0.002) and NF-Del (p=0.009). No significant difference in RFS was observed between NF-Amp and NF-Del (p=0.645) (figure 3B).
Molecular subtypes within insulinomas
Compared with Ins-Amp, Ins-Neutral was significantly smaller (figure 4A). Ins-Amp and Ins-Neutral did not have significant differences in age, gender or G1:G2 ratio (figure 4B, online supplementary table S3). Consistent with the study of chromosome alterations in insulinomas by Wang et al,9 we observed that three of four insulinomas with the loss of chromosome 11 were in the Ins-Neutral, which harboured the CDKN1C. Meanwhile, those with amplification of chromosome 7 were exclusively in the Ins-Amp (48/55), except for one in the Ins-Neutral. CDKN2A deletion was reported to be involved in the metastasis of NF-PanNET30; we evaluated whether CDKN2A also contributed to the pathogenesis of insulinomas. CDKN2A deletion was found in only one Ins-Neutral case, while it was amplified in 47 Ins-Amp and 7 Ins-Neutral cases. No somatic mutations of CDKN2A were detected. No significant associations were found between CDKN2A amplification status and tumour size, histological grade or Ki-67 staining in the insulinomas cohort (see online supplementary table S4). We found that the YY1 mutation rate was significantly higher in Ins-Neutral (17/29) than in Ins-Amp (4/55, p<0.001) (figure 4C), which may explain why YY1 mutations presented exclusively in PUMCH patients as only 1/8 ICGC insulinomas were Ins-Neutral. Previous studies have demonstrated that YY1 T372R mutations regulate the mitochondrial genes IDH3A and UCP2,8 and insulin secretion by increasing the expression of ADCY1 and CACNA2D2 expression.10 Our data showed significantly higher expression levels of UCP2 and ADCY1 in YY1-mutated insulinomas than in YY1-wild-type insulinomas (p<0.001, online supplementary figure S3A,B), based on 44 insulinomas RNA-sequencing data. Given that Ins-Amp had a low YY1 mutation rate, we speculated that early amplified chromosomes drove the tumourigenesis of Ins-Amp. We screened for mTOR-related genes31 on the chromosomes that were most commonly amplified early and found that RHEB had the highest amplification rate (48/84). Meanwhile, we found 47 Ins-Amp with TSC1 amplifications and 15 with TSC2 amplifications (figure 4D). The TSC1–TSC2 complex has inhibitory effects on the function of Rheb. MTOR activity, which was represented by the p-S6 IHC score, was significantly higher in Ins-Neutral than Ins-Amp or NF-PanNETs (figure 4E and F). However, the p-S6 score was not significantly different between YY1-mutant and YY1 wild-type insulinomas (see online supplementary figure S3C).

Molecular subtypes within insulinomas. (A) Comparison of tumour diameters between Ins-Neutral and Ins-Amp. (B) Comparison of histological grades between Ins-Neutral and Ins-Amp. (C) Comparison of YY1 mutation rates between Ins-Neutral and Ins-Amp. (D) Rates of amplification events on early amplified chromosomes involved in mTOR-related genes. (E) IHC p-S6 scores in Ins-Neutral, Ins-Amp tumours and NF-PanNETs. (F) Demonstration of IHC p-S6 staining, with scores of 80, 40 and 0. *P<0.05. IHC, immunohistochemistry; Ins-Amp, insulinoma amplification subtype; Ins-Neutral, insulinoma neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; NS, not significant.
Molecular subtypes within NF-PanNETs
NF-Neutral was significantly smaller in tumour size and higher G1:G2 and N0:N1 ratios compared with NF-Del and NF-Amp (figure 5A–C, online supplementary table S5). Similar trends were observed in both PUMCH NF-PanNETs and ICGC NF-PanNETs (see online supplementary figure S4).

Molecular subtypes within NF-PanNETs. (A) Tumour diameters of the NF-PanNET. (B) The distribution of histological grades in the NF-PanNETs. (C) The presence of lymph node metastases in the NF-PanNETs. (D) Comparison of TMB among NF-PanNET CNV groups. (E) TSG two-hit inactivation in NF-PanNETs. *P<0.05, **P<0.01, ***P<0.001. CNV, copy-number variation; NF-Amp, NF-PanNET amplification subtype; NF-Del, NF-PanNET deletion subtype; NF-Neutral, NF-PanNET neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; NS, not significant; TMB, tumour mutation burden; TSG, tumour suppressor gene.
We subsequently sought to identify potential genetic differences among three CNV subtypes. TMB was significantly higher in NF-Del than in NF-Neutral or NF-Amp as determined from WGS data (figure 5D). With regard to frequently mutated genes, MEN1 and mTOR-related genes31 showed significantly lower mutation rates in NF-Neutral than in NF-Del. The mutation rates of DAXX/ATRX were significantly lower in NF-Neutral compared with both NF-Del and NF-Amp (see online supplementary table S5). Loss of heterozygosity (LOH) of tumour suppressor gene (TSG) PHLDA3 played a role in tumourigenesis of PanNETs.32 Frequent genomic deletion events in the current study prompted us to screen CNVs for LOH in TSGs.33 Overall, all NF-Del and 62% of NF-Neutral have at least one LOH TSG, while 37% of NF-Amp had LOH TSGs. Furthermore, we examined whether the remaining alleles of those LOH TSGs were also inactivated. Collectively, we found that 20 LOH TSGs were coupled with truncating mutations, suggesting the two-hit inactivation of these genes (figure 5E). The most frequently inactivated genes were MEN1 (16 in NF-Del and 9 in NF-Neutral) and DAXX (9 in NF-Del and 4 in NF-Neutral). Inactivation of TSGs was significantly more frequently detected in NF-Del (74%) than in NF-Neutral (25%, p<0.01) and NF-Amp (5%, p<0.01).
Clinical significance of CNV patterns and DAXX/ATRX mutation status
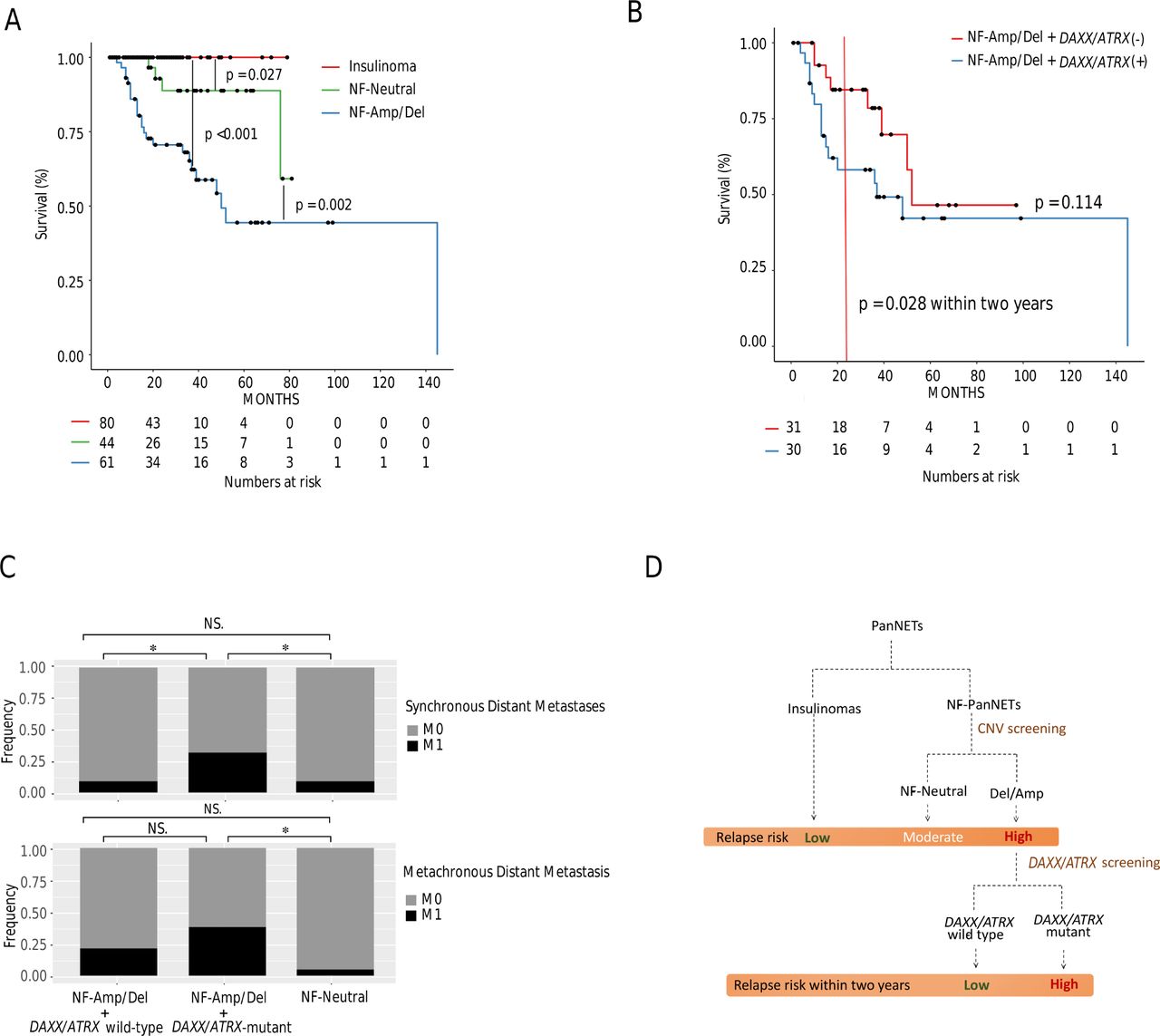
Given that NF-Amp and NF-Del had similar RFS and clinicopathological parameters, these two subtypes were combined as NF-Amp/Del. The RFS of insulinoma was significantly better than that of NF-Neutral (p=0.027) and that of NF-Neutral was significantly better than that of NF-Amp/Del (p=0.002) (figure 6A). In the univariate analysis of RFS in NF-PanNETs, histological grade, lymph node status, distant metastasis, DAXX/ATRX mutations and CNV Del/Amp were significantly associated with shorter RFS (see online supplementary table S6). Since the majority of DAXX/ATRX mutations were in NF-Amp/Del (38/72) and minority in NF-Neutral (6/55, p<0.001), we further compared RFS and clinicopathological parameters in NF-Amp/Del with or without DAXX/ATRX mutations. The RFS showed no significant difference in NF-Amp/Del with or without DAXX/ATRX mutations (p=0.114). However, NF-Amp/Del with DAXX/ATRX mutations had a significantly higher risk of relapse in the first 2-year period (p=0.028, figure 6B, online supplementary figure S5). Irrespective of DAXX/ATRX mutation status, NF-Amp/Del showed shorter RFS, larger tumour size and more advanced histological grade compared with NF-Neutral (see online supplementary table S7). For distant metastasis, NF-Amp/Del with DAXX/ATRX mutations showed more synchronous distant metastases, yet similar rates of metachronous distant metastases, compared with NF-Amp/Del without DAXX/ATRX mutations (figure 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical significance of CNV pattern and DAXX/ATRX mutation status. (A) RFS among three prognostic subtypes. (B) RFS between NF-Amp/Del with or without DAXX/ATRX mutations. (C) The presence of synchronous and metachronous distant metastases among NF-Neutral and NF-Amp/Del with or without DAXX/ATRX mutations. (D) Clinically oriented risk stratification diagram. *P<0.05. CNV, copy-number variation; NF-Amp/Del, NF-PanNET amplification and deletion subtypes; NF-Neutral, NF-PanNET neutral subtype; NF-PanNET, non-functional pancreatic neuroendocrine tumour; NS, not significant; RFS, relapse-free survival.
Discussion
The current study identifies features of SNV and CNV patterns via WGS and/or WES profiling of insulinomas and NF-PanNETs, revealing a new molecular classification system that is suitable for both insulinomas and NF-PanNETs. To the best of our knowledge, this study has an unparalleled scale in regard to patients’ number of the two most common types of PanNETs. PUMCH data were combined with those from the ICGC database using an up-to-date analysis pipeline to ensure valid SNV and CNV calling. We comprehensively compared genetic differences between insulinomas and NF-PanNETs and found the following major features. First, five molecular subtypes are proposed based on three CNV patterns. All CNV patterns (amplification, copy neutral and deletion) are found in NF-PanNETs, while insulinomas lack CNV deletion. Second, SNV mutations exhibit preferences in particular CNV subtypes. YY1 mutations are predominant in Ins-Neutral rather than Ins-Amp. DAXX/ATRX mutations are preferentially observed in NF-Amp and NF-Del, but rarely in NF-Neutral. Third, particular patterns of chromosomes are more likely to be early amplified in Ins-Amp and NF-Amp, yet the early amplified chromosome patterns are different between the two subtypes by Palimpsest calculations. Fourth, the two-hit inactivation of TSG rates vary in different subtypes, and the highest rate is observed in NF-Del.
Scarpa et al 12 recently conducted WGS over a hundred PanNETs and revealed important information regarding the genetic landscape of PanNETs. In their study, the majority of PanNETs were NF-PanNETs and insulinomas accounted for only up to 1/10 of their cases. The genetic landscape of insulinomas is urgently needed. A recent nationwide study led by our centre revealed that approximately 30% of surgically resected PanNETs were insulinomas.34 Our centre is also a high-volume surgical centre for PanNETs.35 36 As a result, we were able to contribute adequate sample sizes for insulinoma sequencing research. Here, PUMCH insulinomas (n=76) consisted of the majority of insulinomas in the combined cohorts. Such sample size is comparable to previous WES studies of insulinomas,9 10 and enable us to provide comprehensive genomic landscape of insulinomas.
The genomic landscape of insulinoma constitutes the basis for the understanding tumourigenesis and pathogenesis. At the SNV mutation level, a previous study by Cao et al 8 revealed approximately 30% (34/113) of insulinomas had detectable YY1 mutations. Our study yields similar YY1 mutation rates in PUMCH insulinomas (27.6%, 21/76) and supports previous findings.8 Furthermore, YY1 mutations were reported to be the driver mutations in insulinomas.8 10 However, it was unknown whether any other driver SNV mutations exist. Here, in addition to WGS, we performed high-depth WES to detect SNV mutations and no other common SMGs were identified. At the CNV level, previous studies showed CNV alterations in various neuroendocrine tumours, such as NF-PanNETs,12 30 37 parathyroid tumours38 and growth hormone secreting pituitary adenomas.39 Our study found CNV amplifications in approximately 65% of insulinomas, which are preferentially found in YY1 wild-type insulinomas (51/63). Moreover, the early chromosome amplifications of insulinomas are more likely to involve the particular chromosomes, such as 7, 3p, 5q and 13q. Molecular pathway alterations by such early amplified chromosomes require further investigation.
Sadanandam et al 7 proposed that the cell origin of NF-PanNETs can be either β-cell precursors or dedifferentiated insulinoma. At the genomic level, previous studies have shown SNV mutational differences between insulinomas and NF-PanNETs8–13; the current study adds new knowledge mainly on the CNV pattern differences between the two types. However, the clue to the cell origin requires transcriptome-based sequencing data. Further investigation is needed to fully elucidate the cell origin of PanNETs.
The current study, in addition to developing more comprehensive molecular classification systems, also investigates their clinical significance. Based on the CNV pattern, we are able to stratify the relapse risk of NF-PanNETs into moderate and high. To the best of our knowledge, this is the first study to comprehensively investigate the prognostic significance of CNV patterns in PanNETs.
It is important to identify molecular markers for relapse predictions and DAXX/ATRX mutations have proven to be effective markers. Loss of DAXX/ATRX expression was associated with reduced survival.40 41 DAXX/ATRX or MEN1 mutations also predicted shorter recurrence-free survival.42 One of the major challenges of DAXX/ATRX mutation-based prediction is that these mutations are found in only 21%–37.5% of NF-PanNETs.12 37 42 The percentage of NF-PanNETs with DAXX/ATRX mutations obtained in the current study is consistent with this percentage (34.6%, 44/127), which limits the sensitivity of tumour relapse predictions. In contrast, we observed that NF-Amp/Del (56.7%, 72/127) can also predict a shorter RFS and contained the majority of DAXX/ATRX-mutated NF-PanNETs. In addition, both NF-Amp/Del with and without DAXX/ATRX mutations showed no significant difference in RFS, which indicated that the prognostic significance of NF-Amp/Del was not entirely dependent on DAXX/ATRX mutations. Given the aforementioned results, we propose that CNV screening could be used to stratify relapse risks. As illustrated in figure 6D, relapse risk can be stratified as low (insulinomas), moderate (NF-Neutral) and high (NF-Amp/Del). Furthermore, NF-Amp/Del with DAXX/ATRX mutations is associated with a higher relapse risk during the first 2-year period. Further clinical studies are needed to validate these findings.
We acknowledge that the current study has several limitations. First, enucleation was proven to be safe for patients with insulinomas in previous studies,36 43 and this surgical procedure was predominantly used for insulinomas resection in the current study. Thus, regional lymphadenectomies and lymph node status estimations for insulinomas might be suboptimal.30 Second, other types of functional PanNETs, such as gastrinomas, glucagonomas and VIPomas, were not included in the current study, and future studies are needed to address these types of PanNETs.
In conclusion, insulinomas and NF-PanNETs have distinctive genetic backgrounds with a novel molecular classification system. A clinically oriented risk stratification diagram for PanNETs has been proposed to facilitate clinical use.
Acknowledgments
We thank Xianze Wang, Peiran Xu and Zhixuan Xuan from Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College for their contributions in clinical data acquisition. We thank Charles J David from Tsinghua University for language editing.
References
Footnotes
XH and SQ are joint first authors.
YZ, KW and WW contributed equally.
Correction notice This article has been corrected since it published Online First. The correspondence details have been updated.
Contributors Study design: WWu, KW, YZ and DL. Study recruitment, clinical sample and data acquisition: XHo, RJ, HC, LL, HD, JJ, TZ, QL, MD, LC, XHa and DG. Bioinformatic analysis: SQ, XHo, FL, ZZ, CY, SL and KW. Statistical analysis: SQ, XHo, FL and RJ. Pathological experiment: WWa, HW, JW, CJ, XL and ZL. Primary results interpretation: XHo, SQ, RJ, LL and WWu. Manuscript drafting: XHo, SQ, RJ, JP, WWu and KW. All authors contributed to and critically reviewed the manuscript.
Funding This work was supported in part by Chinese Academy of Medical Sciences Initiative for Innovative Medicine (CAMS-I2M) 2017-I2M-1-001; National Natural Science Foundation of China (NSFC 81573009, 81773292, 81603157), PUMCH Science Fund for Junior Faculty (JQ201507, pumch-2016-2.22). Science, Technology and Innovation Commission of Shenzhen Municipality under grant no. JCYJ20160531193931852.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval Institutional Review Board of Peking Union Medical College Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.