Article Text

Abstract
Objective Hepatocellular carcinoma (HCC) is a heterogeneous tumour displaying a complex variety of genetic and epigenetic changes. In human cancers, aberrant post-transcriptional modifications, such as alternative splicing and RNA editing, may lead to tumour specific transcriptome diversity.
Design By utilising large scale transcriptome sequencing of three paired HCC clinical specimens and their adjacent non-tumour (NT) tissue counterparts at depth, we discovered an average of 20 007 inferred A to I (adenosine to inosine) RNA editing events in transcripts. The roles of the double stranded RNA specific ADAR (Adenosine DeAminase that act on RNA) family members (ADARs) and the altered gene specific editing patterns were investigated in clinical specimens, cell models and mice.
Results HCC displays a severely disrupted A to I RNA editing balance. ADAR1 and ADAR2 manipulate the A to I imbalance of HCC via their differential expression in HCC compared with NT liver tissues. Patients with ADAR1 overexpression and ADAR2 downregulation in tumours demonstrated an increased risk of liver cirrhosis and postoperative recurrence and had poor prognoses. Due to the differentially expressed ADAR1 and ADAR2 in tumours, the altered gene specific editing activities, which was reflected by the hyper-editing of FLNB (filamin B, β) and the hypo-editing of COPA (coatomer protein complex, subunit α), are closely associated with HCC pathogenesis. In vitro and in vivo functional assays prove that ADAR1 functions as an oncogene while ADAR2 has tumour suppressive ability in HCC.
Conclusions These findings highlight the fact that the differentially expressed ADARs in tumours, which are responsible for an A to I editing imbalance, has great prognostic value and diagnostic potential for HCC.
- Cancer
- Molecular Carcinogenesis
- Gene Expression
- Gene Regulation
- Molecular Pathology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
RNA editing is a widespread post-transcriptional process contributing to greater cellular transcriptome diversity in eukaryotes.
-
In humans, the most frequent type of editing is the conversion of A to I, which is catalysed by the dsRNA specific ADAR family of RNA editing enzymes.
-
Until now, only a few recoding RNA editing events (eg, Q/R site editing in the glutamate receptor) have been verified, and no apparent causal relationship between altered RNA editing levels and cancer progression exists.
-
Accumulating evidence has indicated that a hypo-editing (editing deficiency) phenotype is found in brain tumours and tumour tissues, such as prostate, lung, kidney and testis, and the hypo-editing phenotype is linked to several cancer phenotypes in paediatric astrocytomas and malignant gliomas. It has also been reported that a gene specific hyper-editing phenotype is found in metastatic lobular breast cancer and acute myeloid leukaemia.
What are the new findings?
-
We provide the first extensive analysis of RNA editing in the human liver cancer transcriptome.
-
HCC, distinct from most cancer types, is neither a hypo- nor a hyper-editing cancer; instead, HCC displays a severely disrupted A to I RNA editing balance induced by the differentially expressed ADARs (ADAR1 and ADAR2).
-
Clinically, the differentially expressed ADARs, which are characterised by ADAR1 overexpression and ADAR2 downregulation in tumours, have great prognostic value and diagnostic potential for HCC.
-
ADAR1 has oncogenic ability while ADAR2 functions as a tumour suppressor in HCC.
How might it impact on clinical practice in the foreseeable future?
-
These findings suggest a widespread occurrence of transcript variation at the single nucleotide level in the human liver transcriptome and highlight a link between the RNA editome imbalance in the forms of defective and excess gene specific RNA editing activity to HCC pathogenesis. Monitoring expression levels of ADARs or the global activity of RNA editing represents a useful early biomarker than can be utilised to detect disorders in HCC even before clinical symptoms become apparent.
Introduction
RNA editing is an integral step in generating the diversity and plasticity of cellular RNA signatures. The best characterised type of RNA editing found in mammals converts C to U (cytosine to uracil) and A to I (adenosine to inosine). In humans, the most frequent type of editing is the conversion of A to I, which is catalysed by the double stranded RNA (dsRNA) specific ADAR (Adenosine DeAminase that act on RNA) family of proteins. Because the translation machinery reads inosine as guanosine (G), the ADARs may recode transcripts, which results in a proteome that is divergent from the genome1–4 and thus modulates the protein sequence and function of several gene products. Editing of a specific adenosine is rarely 100% efficient; consequently, the ADARs can generate different protein isoforms in the same cell. Due to the diverse impact of RNA editing on gene expression and function, it is possible that the misregulation of RNA editing may play a role in tumorigenesis by either inactivating tumour suppressor or activating genes that promote tumour progression.
Hepatocellular carcinoma (HCC) is one of the most common types of cancer; it is the third most common cause of cancer related deaths worldwide and it is associated with a poor clinical outcome.5 Furthermore, the incidence of HCC is continually increasing in the USA and Western Europe.6 As with many other solid tumours, HCC development is believed to be a multistep process involving the accumulation of genetic and epigenetic alterations.7 ,8 The recent advent of RNA sequencing (RNA-Seq) has provided a powerful tool that can be used to study changes in transcriptomes and genomes.9–11 In this study, we used RNA-Seq to identify post-transcriptional editing events in three matched pairs of patient derived HCC clinical specimens and their adjacent non-tumour (NT) liver tissue counterparts. We identified an average of 20 007 inferred A to I RNA editing events in non-coding genes and introns, untranslated regions (UTRs) and coding regions of protein coding genes. Because the editing sites occur in coding regions and may result in amino acid substitutions affecting protein properties and interactions, we were particularly interested in the identification and characterisation of RNA editing events within coding regions; these events may be involved or responsible for HCC initiation and progression. Our recent study has reported that the hyper-editing of a gene called antizyme inhibitor 1 (AZIN1) predisposes to human HCC.12 In this study, we performed an extensive analysis of RNA editing in the human liver cancer transcriptome and demonstrated that RNA editing differs between HCC and matched NT liver tissues as follows: (1) unlike most cancer types, HCC displays a disrupted A to I RNA editing balance that is characterised by gene specific hypo-editing and hyper-editing; (2) ADAR1 and ADAR2 but not ADAR3 are responsible for the disrupted editing balance in HCC through their differential expression in HCC compared with NT liver tissues; (3) patients with ADAR1 overexpression and ADAR2 downregulation in tumours demonstrated an increased risk of liver cirrhosis and postoperative recurrence and had poor prognoses; (4) specific recoding events in two genes, FLNB (filamin B, β) and COPA (coatomer protein complex, subunit α) which display the altered editing patterns in tumours compared with normal tissues; and (5) ADAR1 has an oncogenic ability while ADAR2 functions as a tumour suppressor in HCC.
Materials and methods
Detailed methods are included in the online supplementary materials and methods.
Clinical samples
Guangzhou cohort
A total of 125 paired human HCC and adjacent NT tissues that were surgically removed, snap frozen in liquid nitrogen (for protein, RNA and DNA extraction) and embedded in a paraffin block (for tissue microarray construction) were obtained from the Sun Yat-Sen University Cancer Centre (Guangzhou, China), along with their associated clinicopathological summaries, between 2002 and 2007.
Shanghai cohort
A total of 46 paired human HCC and matched NT specimens were obtained from the hepatectomy specimen archives at the Eastern Hepatobiliary Surgery Hospital (Shanghai, China). None of these patients received preoperative chemotherapy or radiotherapy.
Healthy human liver tissues were obtained from donor livers that had not been used for transplantation; the tissues were provided by Dr Man K and Dr Lo CM (Department of Surgery, University of Hong Kong). All of the samples were immediately frozen in liquid nitrogen or fixed in 10% formalin for paraffin embedding. All of the patients provided written informed consents for the use of their clinical specimens for medical research. All of the samples used in this study were approved by the committees for ethics review for research involving human subjects at Sun Yat-Sen University, University of Hong Kong and National University of Singapore.
Cell lines
The SNU-423, 449, 475, 182, 387 and 398 cell lines were obtained from the American Type Culture Collection. All of these cells were maintained in RPMI medium (Gibco BRL, Grand Island, New York, USA) supplemented with 10% fetal bovine serum (Gibco BRL). All of the cell lines used in this study were regularly authenticated by morphological observation and tested for mycoplasma contamination (MycoAlert, Lonza Rockland, Rockland, Maine, USA). The cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Discovery of RNA editing sites
Briefly, aligned files were processed with SAMtools and subsequently VarScan (V.2.2)13 for the detection of A to G (positive strand) or T to C (negative strand) substitutions. Variance calling was constrained to locations within gene regions containing at least 10× coverage, a variation frequency of greater than 10% and a base quality of more than 15. We first filtered the editing sites against the known sites in the NCBI dbSNP database (Build 135) to eliminate germline variants. Second, we eliminated single nucleotide variations (SNVs) for which more than two types of nucleotide sequences were found because these likely represent false positives. Finally, we excluded polymorphic sites with a variation frequency of 100% because they may have resulted from intrinsic mapping errors. After validation, the false positive rate of RNA editing site detection was approximately 40%.
Analysis of RNA editing
Direct sequencing was performed on PCR products, and editing was calculated with the Discovery Studio Gene (DSGene) 1.5 programme (Accelrys Inc, San Diego, California, USA). The reliability of this method was further verified by cloning of individual sequences. PCR products were subcloned into the T-easy vector (Promega), and approximately 50 individual plasmids were sequenced for each sample. The percentage of edited clones was determined and compared with the DSGene quantification. For each sample, 2–3 independent RT-PCR reactions were performed.
Statistical analyses
Unless otherwise indicated, the data are presented as the mean±SD of three independent experiments. The SPSS statistical package for Windows (V.16; SPSS) was used to perform the data analyses. The clinicopathological features of patients with a different status for the differential expression of ADAR1 and ADAR2 were compared using a non-parametric crosstabs analysis (χ2 test) for categorical variables. ADAR1 or ADAR2 expression levels in any two groups of clinical samples (eg, tumours and matched NT liver tissues) were compared using the Mann–Whitney U test. Kaplan–Meier plots and log rank tests were used for disease free survival (DFS) analysis. DFS times were calculated from the data of curative surgery to HCC recurrence, death or the last follow-up data. For the tissue microarray (TMA) analysis, which was based on immunohistochemical (IHC) scores, ADAR1 expression levels in the primary HCC tissues and their matched NT liver tissues were compared using a Wilcoxon signed rank test. A paired Student's t test was used to compare editing levels of FLNB or COPA in HCC and matched NT liver specimens of patients from the Guangzhou (GZ) and Shanghai (SH) cohorts. Editing levels of FLNB and COPA between two preselected groups were compared using the Mann–Whitney U test. An unpaired two tailed Student's t test was used to compare the number of foci, number of migrative and invasive cells, tumour volume and the relative expressions of target genes between any two preselected groups. A p value of <0.05 was considered to be statistically significant.
Results
Global identification of potential A to I editing sites by RNA-Seq
The high throughput transcriptome sequencing (RNA-Seq) of three pairs of primary HCC and matched NT liver tissues (HCC448N/T, HCC473N/T and HCC510N/T) from HCC patients of Chinese origin (GZ cohort) generated 132.3 million reads that could be uniquely aligned to the human genome (hg19). The aligned reads provided substantial coverage (an average of 83.03%) for the vast majority of the identified mRNA transcripts.12 The global identification of potential A to I (G) editing sites was called using VarScan,13 with the following parameters: a minimum coverage depth of 10, a variation frequency of more than 10% and a base quality of more than 15 (figure 1A). To facilitate the detection of actual A to I (G) editing events, we first filtered the editing sites against known events in the NCBI dbSNP database (Build 135) to eliminate germline variants. Next, we eliminated SNVs for which more than two types of nucleotide sequence were found because they likely represented false positives. Finally, we excluded polymorphic sites with a variation frequency of 100% because they may have resulted from an intrinsic mapping error. The distribution of the remaining editing sites in each functional category (coding sequence, UTR, intron, splicing sites, intergenic region and pseudo/ncRNA) is shown in figure 1B and in online supplementary table S1. The majority (approximately 80%) of the inferred editing sites in each of the samples had a low editing level, which ranged from 0% to 20% (see online supplementary figure S1A). Functional enrichment analysis was performed on the genes edited in all three tumours or three NT liver specimens. In both NT liver and tumour samples, the edited genes were found to be most significantly enriched with acute inflammatory response, metabolic process, protein processing and response to wound (see online supplementary tables S2 and S3).

Global identification of potential A to I (G) editing sites by RNA sequencing (RNA-Seq). (A) Distribution of potential editing sites across all of the chromosomes (shown by exterior circle) in three paired hepatocellular carcinoma (HCC) and matched non-tumour (NT) liver samples (HCC448N/T, HCC473N/T and HCC510N/T). The green and purple blocks in the inner circles (deep gray) indicate the A to G substitutions in the tumour and matched NT liver samples, respectively. The green and purple blocks in the inner circle (light gray) indicate the T to C substitutions in the tumour and matched NT liver samples, respectively. UTR, untranslated region. (B) Number of editing sites distributed in each functional category. (C) One example of the UTP14C gene with multiple edits. The RNA editing sites identified from the RNA-Seq data are highlighted by the red boxes. (D) Sequences of individual reads were aligned to the published human genomic sequence of the FLNB (filamin B, β) gene. An A to G conversion was found in the HCC473T sample. The editing and reference events are highlighted by yellow shading. The green boxes denote the editing site reported by DARNED.14 A sequence logo representation of the editing event in the tumour sample is shown below. The height of each letter is proportional to its frequency. (E) Venn diagram illustrating the numbers of exonic editing events which were classified into the three indicated categories. (F) Sequence chromatograms of the AZIN1, FLNB, COPA and UTP14C transcripts in the indicated tumour and matched NT liver samples. An arrow indicates the editing position. The sequence chromatograms of the matching genomic DNA (gDNA) sequences of each gene are shown in online supplementary figure S2.
To experimentally validate our calls, we verified a subset of potential sites by performing Sanger sequencing of both DNA and RNA from the same samples that were utilised for RNA-Seq. We validated 30 inferred editing sites, and the majority (21/30, 70%) were verified (see online supplementary figure S1B). Notably, UTP14C mRNA contains previously undescribed A to G substitution at multiple sites (figure 1C). We also compared sites identified in this study with editing sites in the DARNED database14 and a human B cell dataset.15 An example of an editing target, FLNB, exhibited confirmed, extensive, non-synonymous editing at the same site as in the human B cell dataset. However, no transcript alteration was identified at the position described in the DARNED database (figure 1D). These results support the current notion that RNA editing frequency can be regulated in a tissue or cell type specific manner.16
HCC displays a disrupted editing balance
To better understand the link between the RNA editing process and hepatocarcinogenesis, we applied several adjustments and additional filters to facilitate the identification of editing sites within coding regions that may have highly tumour suppressive or oncogenic potentials. When the coverage depth was not less than 15, the editing sites in the coding regions detected in more than one NT liver but not in HCC tissues were assigned to the ‘NT specific editing’ category (figure 1E and see online supplementary table S4); conversely, those sites found in more than one tumour tissue but not in NT liver specimens were placed in the ‘tumour specific editing’ category (figure 1E and see online supplementary table S5). The editing sites that were found in more than one pair of HCC and matched NT liver tissues were placed in the ‘common editing events’ category (figure 1E and see online supplementary table S6). Interestingly, two recoding editing sites in AZIN112 and FLNB, which are in the ‘common editing events’ category, demonstrated higher A to I (G) editing frequencies in HCC448T and HCC473T than in HCC448N and HCC473N, respectively (figure 1F and online supplementary figure S2). As a member of the ‘NT specific editing’ category, an A to I (G) editing site at codon 164 (Ile→Val) of the COPA gene was completely absent in all three tumour samples (figure 1F and see online supplementary figure S2). Conversely, a ‘tumour specific’ editing site within the UTP14C coding region only exhibited an A to G substitution in the tumour samples (figure 1F and see online supplementary figure S2). These results suggest that HCC is neither a hypo-editing nor a hyper-editing cancer; instead, HCC displayed a severely disrupted A to I RNA editing balance.
In the past few years, bioinformatic and experimental studies have revealed that the A to I editing events occur in non-coding repetitive sequences, mostly Alu elements, and tend to undergo multi-editing in tight clusters.17 To date, it is commonly accepted that a reduced A to I editing in general may be involved in the pathogenesis of cancer, and a significant global hypo-editing of Alu repetitive elements was observed in brain, prostate, lung, kidney and testis tumours.17 In our study, the numbers of potential A to I (G) editing sites within the Alu sequences in three tumours were higher than their corresponding NT liver samples (see online supplementary figure S3A). We validated 30 editing sites within Alu sequences and all were verified. The editing levels of two editing sites within the Alu repetitive element of the gene TTPA (tocopherol (α) transfer protein) were dramatically decreased in tumour samples (see online supplementary figure S3B). In contrast, five editing sites in Alu sequences of two genes called MAGT1 (magnesium transporter 1) and PAICS (phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succinocarboxamide synthetase) were highly edited in tumour samples (see online supplementary figure S3B). All of these data suggest that there is a disrupted A to I editing balance in coding regions and non-coding Alu repetitive elements in human HCC.
Differentially expressed RNA editing enzyme ADAR1 and ADAR2 in HCC
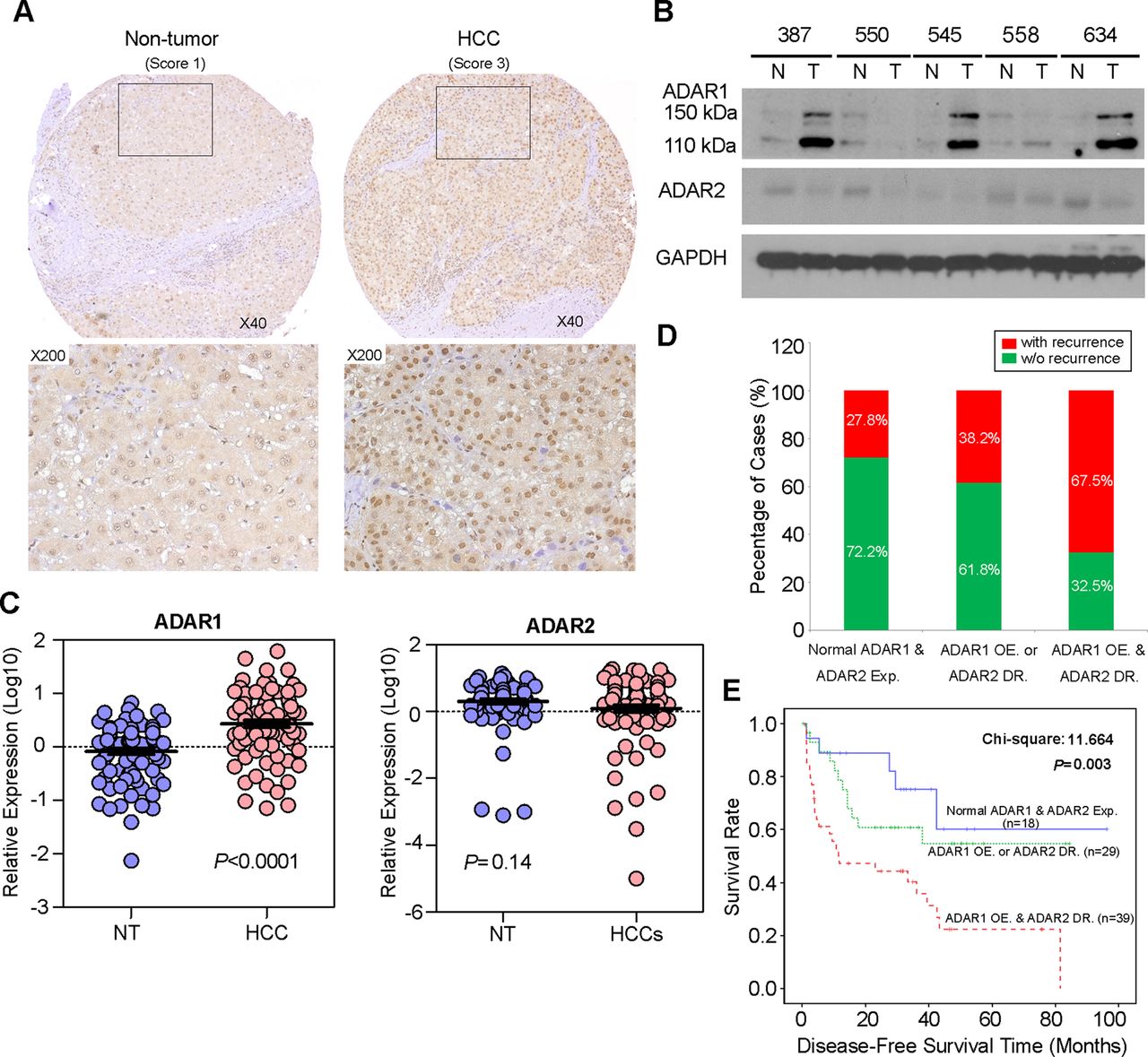
A to I RNA editing is a post-transcriptional modification in stem loop structures within precursor mRNA, which is catalysed by dsRNA specific ADAR enzymes.18 In humans, the ADAR family is composed of three independent genes, ADAR1–3. ADAR1 and ADAR2 are expressed in many tissues whereas ADAR3 is specifically expressed in the brain.3 As described previously, RNA-Seq profiling of the ADARs indicated that two ADAR1 transcript variants (NM_001025107 and NM_015840) encoding 110 kDa (p110) and 150 kDa (p150) isoforms, respectively, demonstrated relatively high abundances in liver tissue.12 However, ADAR2 and ADAR3 were expressed at extremely low levels and were undetectable in all samples.12 In this study, we constructed a panel of TMAs consisting of 92 surgically resected primary HCCs and their matched NT liver tissues from the GZ cohort. By performing IHC staining, we observed the differential nuclear expression of ADAR1 between the primary HCC and matched NT liver tissues. A detailed analysis of the IHC data revealed the ADAR1 was overexpressed in 71.7% (66/92) of the analysed HCC tissues (p<0.001, Wilcoxon signed rank test) (figure 2A and see online supplementary table S7). However, it was found that the sensitivity of IHC was too low to detect ADAR2 expression in both the primary HCC and matched NT liver tissues. Therefore, we determined expression of ADAR2 and ADAR1 using a western blot analysis and found that ADAR2 expression in approximately 50% (15/30) of the HCC samples was lower than in the matched NT liver specimens (figure 2B). Consistently, ADAR1 protein expression (both p110 and p150 isoforms) in approximately 73% (22/30) of the HCC specimens was higher than in the matched NT liver specimens (figure 2B). In order to obtain the expression levels of both ADAR1 and ADAR2 in all 92 paired HCCs and their matched NT liver tissues, we subsequently used quantitative real time PCR to examine ADAR1 and ADAR2 expression in 92 paired specimens that were utilised for TMA construction. As a result, ADAR1 was significantly overexpressed in the HCC specimens compared with the NT liver specimens (p<0.0001, Mann-Whitney U test) (figure 2C). However, ADAR2 expression was obviously decreased in approximately 47% (43/92) of the tumour samples compared with the NT samples (p=0.14, Mann-Whitney U test) (figure 2C).

Differential expression of ADAR (Adenosine DeAminase that acts on RNA) 1 and ADAR2 in hepatocellular carcinoma (HCC) and its clinical implication. (A) Example of ADAR1 expression level detected in a primary HCC tumour and its matched non-tumour (NT) liver specimen. Based on staining intensities, ADAR1 immunoreactivities were scored as strong expression (3) and weak (1) expression in the primary HCC and matched NT liver specimens, respectively (see the online supplementary materials and methods section for details). The boxed regions are magnified and displayed in the lower panels. (B) Western blot analyses of ADAR1 and ADAR2 expression levels in five paired HCC and matched NT liver specimens. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (C) Dot plots represent relative ADAR1 (left) and ADAR2 (right) expression levels in HCC and corresponding NT liver tissue samples, as detected by quantitative real time PCR (mean±SD, n=92; Mann–Whitney U test). (D) Association between the postoperative recurrence rate and status of expression of ADARs (p=0.004, χ2 test). (E) Kaplan–Meier plots for disease free survival rate of patients demonstrating ADAR1 overexpression (OE) and ADAR2 downregulation (DR) (red line; n=39), ADAR1 OE or ADAR2 DR (green line; n=44) and normal ADAR1 and ADAR2 expression in tumours (blue line; n=23) (log rank test).
Based on the quantitative real time PCR analysis of ADAR1and ADAR2 expression, patients with ADAR1 overexpression (defined as a twofold increase in ADAR1 expression in tumours) and ADAR2 downregulation (defined as a twofold decrease in ADAR2 expression in tumours), demonstrated higher incidences of tumour recurrence (p=0.004) and liver cirrhosis (p=0.016) and shorter DFS times (p = 0.003) than patients who had ADAR1 overexpression or ADAR2 downregulation and patients who had neither ADAR1 overexpression nor ADAR2 downregulation (figure 2D, E and table 1). In the univariate Cox analyses, the statistically significant predictors for a patient's DFS were liver cirrhosis, American Joint Committee on Cancer tumour staging and the differentially expressed ADARs in tumours (see online supplementary table S8). In the multivariate Cox analyses, differentially expressed ADARs in tumours were shown to be an independent prognostic factor for DFS (p=0.025, HR 1.725; 95% CI 1.071 to 2.777; see online supplementary table S8). We conclude that the differentially expressed ADAR1 and ADAR2 in HCC, as shown by ADAR1 overexpression and ADAR2 downregulation in tumours, predicts a poor prognosis for HCC patients.
Clinicopathological analyses of the differentially expressed ADARs in the Guangzhou cohort of 92 primary hepatocellular carcinoma patients
Differentially expressed ADARs contribute to the altered gene specific editing patterns in HCC
Due to the differentially expressed ADARs, the A to I editing balance could be disrupted in HCC. In this study, we were particularly interested in two exemplary editing targets, FLNB and COPA. To clarify which ADARs are responsible for FLNB and COPA editing, the tumour samples were divided into ‘high level’ and ‘low level’ groups based on the average relative quantification (RQ) values of ADAR1 or ADAR2 in all 92 tumour specimens (Avg[ADAR1]: 5.84; Avg[ADAR2]: 4.98). Tumours with ‘high level’ ADAR1 expression (RQ≥5.84) demonstrated a significantly higher editing level of FLNB (p=0.003) but not COPA (p=0.81; Mann–Whitney U test) (figure 3A). Tumours with ‘high level’ ADAR2 expression (RQ≥4.98) were found to have higher editing degrees of both COPA (p=0.013) and FLNB (p=0.091; Mann–Whitney U test) (figure 3B). The ADAR1 p150 isoform is presumably responsible for the A to I editing of viral RNAs produced by viruses18 ,19 but not of the nuclear pre-mRNAs.20 To directly determine whether ADARs regulate FLNB and COPA editing, either the ADAR1 p110 isoform or ADAR2 was overexpressed in the HCC cell line SNU-423. SNU-423 cells overexpressing ADAR1 p110 displayed enhanced FLNB editing whereas ADAR1 overexpression did not affect COPA editing (figure 3C). The introduction of ADAR2 into SNU-423 cells resulted in increased COPA and FLNB editing levels (figure 3D). To further confirm our findings, we conducted knockdown/rescue experiments with SNU-423 cells that stably expressed ADAR1 p110 (423-AR1) or ADAR2 (423-AR2). Silencing of ADAR1 expression using a small hairpin RNA (shRNA) against ADAR1 (shADAR1) in 423-AR1 cells dramatically decreased the FLNB editing level from 27.4% to 8.2%; this editing was effectively rescued by overexpressing an ADAR1 p110 mutant that preserves the native amino acid sequence but contains six point mutations within the ADAR1 shRNA targeting site (figure 3E,G). Using the same strategy, ADAR2 knockdown in 423-AR2 cell lines dramatically decreased the editing levels of both FLNB and COPA, and the editing was effectively abolished by reintroducing the ADAR2 mutant into 423-AR2 cells (figure 3F, G). Together, these results likely suggest that the differential expression of ADAR1 and ADAR2 in tumours, which is tightly associated with the altered gene specific editing pattern, may explain the A to I editing imbalance in HCC.

FLNB (filamin B, β) editing is catalysed by both ADAR (Adenosine DeAminase that acts on RNA) 1 and ADAR2, while COPA (coatomer protein complex, subunit α) editing is specifically catalysed by ADAR2. (A, B) FLNB and COPA editing level in tumours with ‘high level’ and ‘low level’ expression of ADAR1 (A) or ADAR2 (B). The data are presented as box plots with median (horizontal line), 25–75% (box) and 5–95% (error bar) percentiles for each group. The mean is indicated as ‘+’ (Mann–Whitney U test) and the dots indicate the outliers. (C, D) Left: Western blot analyses of ADAR1 and ADAR2 expression in SNU-423 cells that were transiently transfected with an ADAR1 p110 variant expression construct (ADAR1 p110) or empty vector (CTL) (C) or SNU-423 cells that were transiently transfected with an ADAR2 expression construct (ADAR2) or empty vector (CTL) (D). (C, D) Right: sequence chromatograms of the FLNB and COPA transcripts in the indicated cell lines. The percentages of edited FLNB or COPA transcripts were detected as described in the Materials and Methods section. An arrow indicates the editing position. (E, F) Following transfection with the indicated constructs into SNU-423 cells that stably expressed the ADAR1 p110 isoform (E) or ADAR2 (F) (see the online supplementary materials and methods section for details), ADAR1 and ADAR2 expression levels were detected by western blot analysis. (G) Editing levels of FLNB and COPA in each cell line as indicated are shown in a bar chart (mean±SD of three independent experiments; *undetectable).
To further investigate the relationship between RNA editing and HCC progression, we also examined the editing frequencies of two editing targets, FLNB and COPA, in healthy human liver tissues (n=8) and two different cohorts of primary HCC and NT liver samples from GZ (n=125) and SH (n=47) cohorts. Two individual cohorts of HCC and matched NT liver samples demonstrated dramatically higher FLNB editing degrees compared with the healthy liver specimens (figure 4A). COPA editing levels were remarkably lower in both the HCC and matched NT liver specimens from two cohorts than those in the healthy liver specimens (figure 4B). Moreover, two individual cohorts of HCC samples displayed significantly higher and lower editing levels of FLNB and COPA, respectively, compared with the matched NT liver tissues (FLNB: pGZ<0.0001, pSH<0.0001; COPA: pGZ<0.0001, pSH<0.0001; Mann–Whitney U test) (figure 4A,B). All of these data suggest that the altered gene specific editing activities are closely associated with HCC pathogenesis from normal to adjacent non-tumour to clinically verified HCC.

Altered gene specific editing patterns induced by the differentially expressed ADARs (Adenosine DeAminases that act on RNA) is closely associated with hepatocellular carcinoma (HCC) pathogenesis. (A, B) Dot plots represent FLNB (filamin B, β) (A) and COPA (coatomer protein complex, subunit α) (B) editing levels in healthy human liver tissues (mean±SD, n=8) and in 125 matched primary HCC and non-tumour (NT) liver samples in the Guangzhou (GZ) cohort and in 47 matched primary HCC and NT liver samples in the Shanghai (SH) cohort (Mann–Whitney U test). (C, D) FLNB (C) and COPA (D) editing levels in HCC and matched NT liver specimens from 125 and 47 patients in the GZ and SH cohorts (paired Student's t test).
Because the average FLNB or COPA editing level was approximately 10% different between the NT liver and tumour specimens in the GZ cohort (FLNB: 18.9 vs 31.6%; COPA: 20.7 vs 7.1%), we set an increase of not less than 10% of the FLNB editing level in tumour tissues when compared with NT liver tissues as the cut-off value for FLNB hyper-editing to subdivide HCC patients. Similarly, a decrease of not less than 10% of the COPA editing level in tumour tissues when compared with NT liver samples was used as the cut-off value for COPA hypo-editing. In the GZ cohort, approximately 52% (65/125) and 67% (84/125) of the HCC specimens displayed FLNB hyper-editing or COPA hypo-editing, respectively compared with the matched NT liver specimens (FLNB: p<0.0001; COPA: p<0.0001; paired Student's t test) (figure 4C,D). Consistently, FLNB hyper-editing and COPA hypo-editing were found in approximately 49% (23/47) and 74% (35/47) of the HCC cases in the SH cohort, respectively (FLNB: p<0.0001; COPA: p<0.0001; paired Student's t test) (figure 4C,D).
We conclude that as a consequence of the differentially expressed ADARs (ADAR1 and ADAR2), HCC displays a disrupted A to I editing balance, which was reflected by the altered gene specific editing patterns.
ADAR1 has oncogenic ability, while ADAR2 functions as a tumour suppressor gene
As upstream regulators of A to I RNA editing, ADAR proteins have a number of reported target transcripts, such as serotonin receptor subunit 2C (5-HT2CR), glutamate receptor (GluRs), filamin A (FLNA) and bladder cancer associated protein (BLCAP).17 ,21 ,22 This prompted us to study the functional roles of ADAR1 and ADAR2 during hepatocarcinogenesis. For this purpose, we introduced ADAR1 (p110) or ADAR2 expression constructs into two HCC cell lines (SNU-423 and SNU-449) expressing the relative low endogenous ADAR1 (p110) and ADAR2 among six HCC cell lines using a lentiviral system (figure 5A,B). As detected by in vitro functional assays, cells transduced with the ADAR1 lentivirus (423-AR1 and 449-AR1) had accelerated growth rates and higher frequency of focus formation than cells transduced with the control LacZ lentivirus (423-LacZ and 449-LacZ) (figure 5C and see online supplementary figure S4A). However, introduction of ADAR2 into SNU-423 and SNU-449 cells (423-AR2 and 449-AR2) could effectively inhibit tumorigenic abilities, including significant inhibition of cell growth rate and reduction in frequency of focus formation (figure 5C and see online supplementary figure S4A). Consistent with the clinical correlation between tumour recurrence and the differentially expressed ADAR1 and ADAR2 in tumours, 449-AR1 and 449-AR2 cells demonstrated increased and decreased migrative and invasive capabilities, respectively, compared with 449-LacZ cells (figure 5D,E). As expected, this phenotype could also be observed in 423-AR1 and 423-AR2 cells compared with control cells (see online supplementary figure S4B,C). Xenograft studies in mice demonstrated that the growth rate of tumours induced by 449-AR1 or 449-AR2 cells was markedly higher or lower, respectively, than those induced by 449-LacZ cells (figure 5F,G). All of these data demonstrate that ADAR1 and ADAR2 have the opposite effects on tumorigenicity. ADAR1 has oncogenic ability while ADAR2 functions as a tumour suppressor gene, suggesting that there is a tight link between the unbalanced A to I editing mediated by the differentially expressed ADARs and HCC initiation and progression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ADAR1 (Adenosine DeAminase that acts on RNA 1) has oncogenic ability while ADAR2 functions as a tumour suppressor gene. (A) Relative ADAR1 p110 and ADAR2 expression levels in six hepatocellular carcinoma cell lines, as detected by quantitative real time PCR (mean±SD of three independent experiments). (B) Western blotting showing expression of ADAR1 p110 and ADAR2 proteins in the indicated cell lines. β-Actin was the loading control. (C) Quantification of foci formation induced by the indicated stable cell lines. Triplicate independent experiments were performed and the data were expressed as the mean±SD of triplicate wells within the same experiment (unpaired two tailed Student's t test). Scale bar 1 cm. (D, E) Quantification of cells from the indicated cells that invaded through the Matrigel coated membrane (D) or migrated through the polyethylene terephthalate membrane (E) (unpaired two tailed Student's t test). Scale bar 200 μm. (F) Growth curves of tumours derived from the indicated cell lines over a period of 8 weeks. Data are presented as mean±SD (unpaired two tailed Student's t test). (G) Volumes of tumours derived from the indicated cell lines at the end point. Data are presented as mean±SD (unpaired two tailed Student's t test). *undetectable.
Discussion
The data from our transcriptome measurement demonstrate differentially expressed gene profiling and reveal multiple types of SNVs. These variants are expected to consist of (in descending order of frequency) inherited polymorphisms, somatic mutations, sequencing errors and actual differences between RNA and DNA (eg, RNA editing and polyadenylation). RNA editing is broadly defined as the post-transcriptional process that alters the sequence of primary RNA transcripts. Of the various types of RNA editing, A to I (G) modification is most widespread in higher eukaryotes.18 ,23 ,24 The most frequent RNA editing mechanism in mammals involves the conversion of specific adenosines into inosines by the ADAR family of enzymes. The majority of A to I substitutions occur not within coding portions of mRNA but largely in non-coding RNAs.25 Early RNA editing studies have revealed that the editing occurs in many tissues and organs. In humans, this process is thought to occur predominantly in the brain and may be a key regulator of neural development. Recent studies have demonstrated that altered RNA editing is associated with numerous human pathologies, particularly cancers. Accumulating evidence has indicated that a hypo-editing phenotype is found in brain tumours and tumour tissues, such as prostate, lung, kidney and testis; additionally, the hypo-editing phenotype is linked to several cancer phenotypes in paediatric astrocytomas and malignant gliomas.17 ,26 ,27
Recent studies have revealed that the developmental and cell type specific modulation of A to I RNA editing is linked to ADAR expression and localisation.19 It has been reported that all of the three editing enzymes, ADAR1, ADAR2 and ADAR3, were found to be downregulated in brain tumours. Consistently, overexpression of ADAR1 and ADAR2 in the U87 glioblastoma multiforme cell line resulted in a decreased proliferation rate, suggesting that the reduced A to I editing in brain tumours is involved in the pathogenesis of cancer.17 In contrast, ADAR1 and/or ADAR2 were found to be upregulated in tumour tissues, such as prostate cancer and breast cancer tissues.28 Similar to many other solid tumours, HCC development is believed to be a multistep process involving the accumulation of genetic and epigenetic alterations.7 ,8 However, the role of RNA editing in HCC progression remains unknown. In this study, we identified an average of 20 007 A to I RNA editing events in transcripts. Intriguingly, unlike most types of cancers that are associated with a general decrease or increase in RNA editing activity, HCC is neither a hypo- nor hyper-editing cancer and displays a disrupted A to I editing balance in coding regions and non-coding Alu repetitive elements in human HCC. Moreover, the connection between the differential expression of ADARs and an altered gene specific editing pattern was investigated for the first time to illustrate how the A to I RNA editing balance was deregulated in HCC. Based on RNA-Seq transcript quantitation, the highest transcript abundance of ADAR1 was found in liver tissue whereas ADAR2 was expressed at extremely low levels, and ADAR3 was undetectable in all samples. Most ADAR proteins localise to the nucleus, with the exception of the ADAR1 p150 isoform which is shuttled between the nucleus and cytoplasm and is thought to be responsible for the A to I editing of the viral RNA produced by viruses in the cytoplasm of infected cells.18 ,20 In this study, we revealed that both of the ADAR1 p110 and p150 variants were overexpressed in approximately 70% of the primary HCC samples whereas ADAR2 was downregulated in approximately 50% of HCC cases. Clinically, the differentially expressed ADAR1 and ADAR2 in HCC, as shown by ADAR1 overexpression and ADAR2 downregulation in tumours, predicts a poor prognosis for HCC patients.
In addition, A to I editing can be very specific, leading to deamination of select adenosine residues, or it can be almost random and lead to non-selective conversion of many inosines. For long dsRNA (>100 bp) within 3′UTR regions, many adenosine residues are edited promiscuously, leading to approximately 50% of adenosines being converted to inosines. However, in terms of A to I editing of protein coding sequences, it is highly selective, and an imperfect fold back dsRNA structure is formed between the exon sequence surrounding the editing site(s) and a downstream intronic complementary sequence termed editing site complementary sequence.24 As described in our recent study, the AZIN1 transcript undergoes A to I editing by a similar mechanism involving the fold back dsRNA structure configured from complementary edited exon 11 and the downstream 100 bp intronic sequences.12 In addition, ADAR1 has a 5′ nearest neighbour preference of A=U>C>G but no reported 3′ nearest neighbour preference.29 On the other hand, ADAR2 has a 5′ nearest neighbour preference of A≈U>C=G and 3′ nearest neighbour preference of U=G>C=A.30 In this study, two representative recoding editing events at codon 164 (Ile→Val) of the COPA gene and codon 2269 (Met→Val) of the FLNB gene were selected for further study. The overexpression and knockdown/rescue experiments demonstrated that FLNB editing was catalysed by both ADAR1 and ADAR2 and that COPA editing was specifically catalysed by ADAR2. Intriguingly, two individual cohorts of HCC samples displayed significantly higher and lower editing levels of FLNB and COPA, respectively, compared with matched NT liver tissues. Moreover, the altered gene specific editing activities were closely associated with HCC pathogenesis from normal to adjacent non-tumour to clinically verified HCC. To our knowledge, unlike the editing events that occur within 3′UTR regions, where the editing can affect transcript stability via affecting microRNA targeting or the nuclear retention of transcripts, those within coding regions will cause amino acid change and affect protein function rather than protein level. Similar to the FLNB transcript, AZIN1 is one of the recoding editing targets that are placed in the ‘common editing events’ category. As a result of the A to I editing of AZIN1 transcripts, the serine (S)→glycine (G) substitution at residue 367, located in β strand 15 (β15) and predicted to cause a conformational change, induced a cytoplasmic to nuclear translocation and conferred gain of function phenotype. Moreover, AZIN1 editing was catalysed by ADAR1, and an 18% increase in AZIN1 editing frequency was sufficient to promote tumorigenic phenotypes,12 strongly suggesting there exists a causative relationship between the altered RNA editing activity and HCC progression. ADAR1 and ADAR2 proteins have numerous editing substrates; although it is tempting to investigate if the editing alterations in the specific genes analysed may be relevant to the malignant phenotype, studying the functional connection between the decreased/increased expression of ADARs and carcinogenesis is of extreme biological importance. Here, our functional studies have indicated that ADAR1 has oncogenic ability while ADAR2 functions as a tumour suppressor gene. Therefore, we propose a model in which the precise regulation of the expression levels of ADARs is essential for accurate editing, and the altered expression of ADARs could be at the origin of cell transformation.
Investigating the connection between RNA editing and cancer progression is only the initial step in this research. Recent efforts to identify RNA editing events in the human transcriptome using deep sequencing approaches have indicated that many of the identified RNA–DNA differences could be explained by errors in sequencing or mapping errors in the assignment of RNA-Seq reads to the reference transcriptome.31–33 Moreover, most recoding sites may be modified only to levels of less than a few percent.19 In this study, we reported two recoding editing events with high level modification rates. Specifically, FLNB editing could be detected in nearly all of the primary HCC samples. More importantly, as a result of the differentially expressed ADARs (ADAR1 and ADAR2) in tumours, alterations in the gene specific editing activities are closely associated with HCC pathogenesis. Therefore, we speculate that monitoring expression levels of ADARs or the global activity of RNA editing represents a useful early biomarker for the detection of disorders in HCC before clinical symptoms become apparent. Generation of in vivo models for gene specific editing deficiency or hyper-editing should better elucidate the physiological significance of particular editing events in the context of liver cancer.
Acknowledgments
We wish to thank those patients who donated tumour tissues to our tissue bank.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 10 - Online table 5
- Data supplement 11 - Online table 6
- Data supplement 12 - Online table 7
- Data supplement 13 - Online table 8
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
- Data supplement 5 - Online figure 4
- Data supplement 6 - Online table 1
- Data supplement 7 - Online table 2
- Data supplement 8 - Online table 3
- Data supplement 9 - Online table 4
Footnotes
-
THMC and CHL contributed equally to this work.
-
THMC and CHL contributed equally to this work.
-
Contributors THMC and CHL were involved in study conception and design, data acquisition, analysis and interpretation, manuscript drafting, and finalisation and submission. CHL performed illumina mRNA library preparation and all bioinformatics analysis of transcriptome sequencing. LQ contributed to data acquisition, analysis and interpretation. YL, KJY, ML, YS, JF, VHEN and RKKC provided technical expertise and support. Y-FY performed patient identification, enrolment and follow-up, and clinical database compilation and analysis. LC developed the study concept, and critically revised and finalised the manuscript with input from X-YG and DGT.
-
Funding This work was supported by the Singapore Translational Research (STaR) Award (NMRC/STaR/0001/2008), a grant from the Singapore Ministry of Education and National Research Foundation, the Hong Kong Research Grant Council Central Allocation (HKU5/CRF/08), the Hong Kong RGC Collaborative Research Grant (HKU 7/CRG09), the ‘Hundred Talents Program’ at Sun Yat-Sen University (85000-3171311) and the National Key Sci-Tech Special Project of Infectious Diseases (2012ZX10002-013).
-
Competing interests None.
-
Ethics approval The study was approved by the committees for ethics review for research involving human subjects at Sun Yat-Sen University, University of Hong Kong and National University of Singapore.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing The RNA-Seq data were deposited in the following repository: Repository/DataBank Accession: GEO; Accession ID: GSE33294. Databank URL: http://www.ncbi.nlm.nih.gov/geo/.
Linked Articles
- Commentary
- Digest